 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库亚纳米Rh-Co双原子协同催化腈类加氢研究取得新进展
仲胺类化合物广泛存在于药物、农用化学品及功能材料中,全球每年胺类产量超过600万吨。腈类催化加氢是合成仲胺的重要途径,但传统贵金属催化剂面临活性与选择性的“跷跷板”困境:一方面易发生过度加氢或偶联副反应,目标仲胺选择性通常仅60-75%;另一方面需要较高温度(>373 K)和压力(3-10 MPa H2)。单原子催化剂虽能通过孤立位点提高选择性,但单一活性中心难以协同活化H2和较大尺寸的腈类底物,导致催化活性不足。
近日,中国科学院金属研究所沈阳材料科学国家研究中心刘洪阳研究员和博士研究生陈家威等,联合重庆大学孙耿副教授、北京大学马丁院士等团队,成功在富缺陷石墨烯载体(ND@G)上构建了原子级分散的Rh-Co异核双原子催化剂(Rh1Co1ND@G),实现了温和条件下苯甲腈的高效加氢制二苄胺。该成果发表于国际学术期刊《Nature Communication》(https://doi.org/10.1038/s41467-026-69778-2),为设计高性能原子级分散加氢催化剂提供了新思路。
研究团队在载体上先锚定Rh原子,再引入Co(NH3)42+配合物,经氢气还原后成功构筑了Rh-Co双原子对位点。高角环形暗场扫描透射电镜(HAADF-STEM)清晰观察到大量相邻的Rh-Co原子对,原子间距约2.55 Å(图1)。电子结构分析表明Rh和Co的电子结构相互调制,表明两者之间存在强电子耦合效应。催化性能测试表明,在333 K、0.6 MPa H2的温和条件下,Rh₁Co₁/ND@G催化苯甲腈加氢的转换频率(TOF)高达4068 h-1,是相应Rh单原子催化剂的1.6倍,对二苄胺的选择性超过98%。该性能优于目前已报道的所有多相催化剂。催化剂在循环使用12次后仍保持85%以上产率,展现了优异的耐久性(图2)。此外,该催化剂对多种含吸电子基或给电子基的芳香腈、杂环腈甚至乙腈均表现出良好的加氢性能,生成相应仲胺的产率优异,展现了广泛的底物适应性。
结合动力学实验、程序升温脱附(TPD)、原位红外光谱及密度泛函理论(DFT)计算,研究团队揭示了Rh-Co双原子位点的协同催化机理(图3)。结果表明,Rh位点主要负责H2的活化与解离,而Co位点通过增强苯甲腈的吸附(尤其是苯环相互作用)优化了底物在双原子位点上的构型。这种协同作用使C≡N键极化,并将加氢决速步的能垒从Rh单原子催化剂的0.81 eV大幅降低至0.61 eV,从而在动力学上显著促进了低温反应速率。图4为Rh₁Co₁/ND@G上催化苯甲腈加氢制二苄胺的反应示意图。
该工作不仅开发了一种高效双原子金属催化剂的制备策略,更从原子尺度揭示了异核双原子协同催化机理,为面向绿色化学与精细化工领域设计高效、稳定的原子级分散加氢催化剂提供了新的思路。
以上工作得到了国家自然科学基金、国家重点研发计划、国家高层次人才项目、中国科学院引进人才计划项目、中国科学院大科学装置建制化项目与中国石化、中国中化等企业合作项目提供的支持,以及上海同步辐射光源、北京同步辐射光源的大力支持。
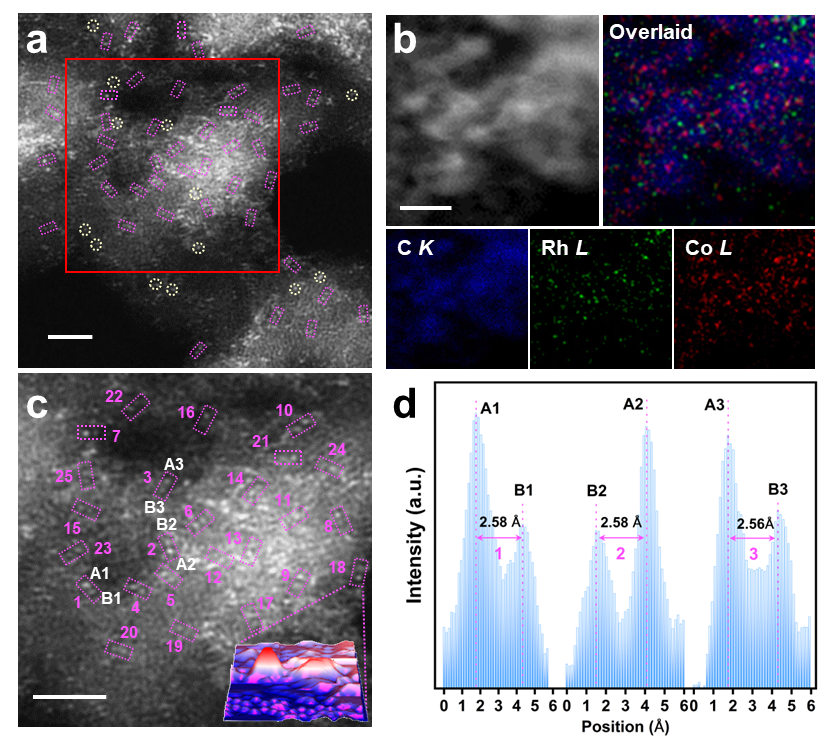
图1.催化剂结构表征。(a-c)HAADF-STEM图像,(d)原子间距统计。
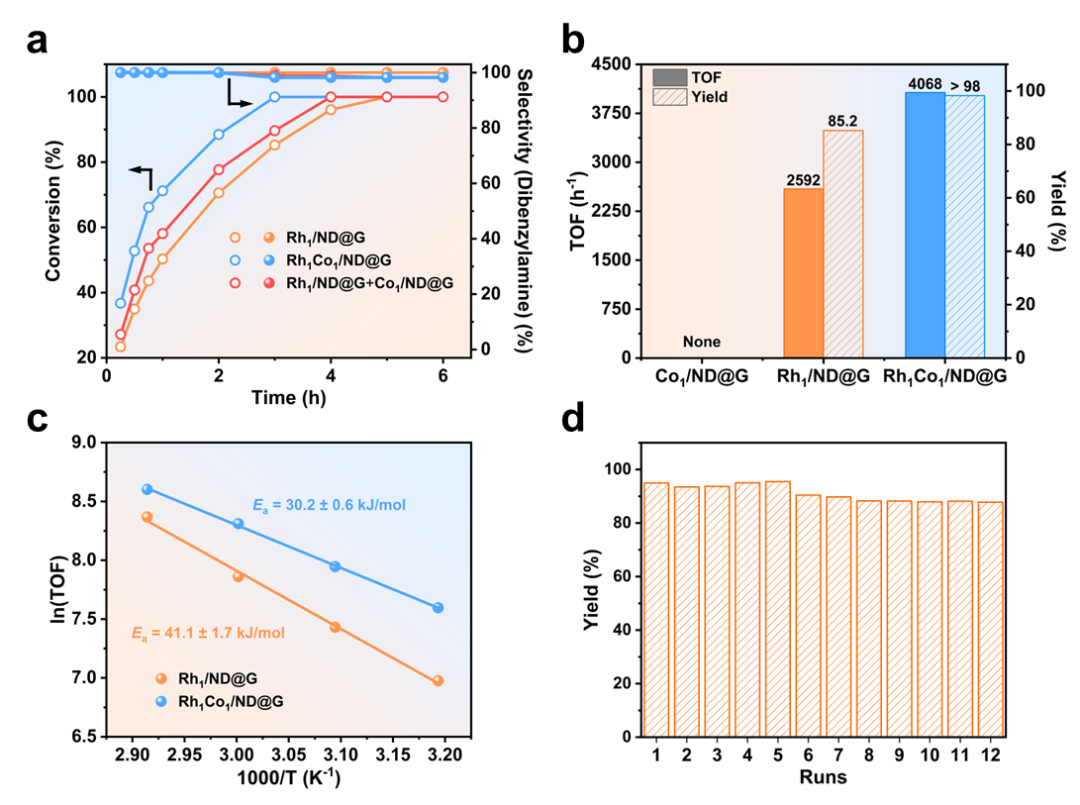
图2. 苯甲腈催化加氢性能。(a)时间-转化率曲线,(b)TOF对比,(c)表观活化能,(d)循环稳定性。
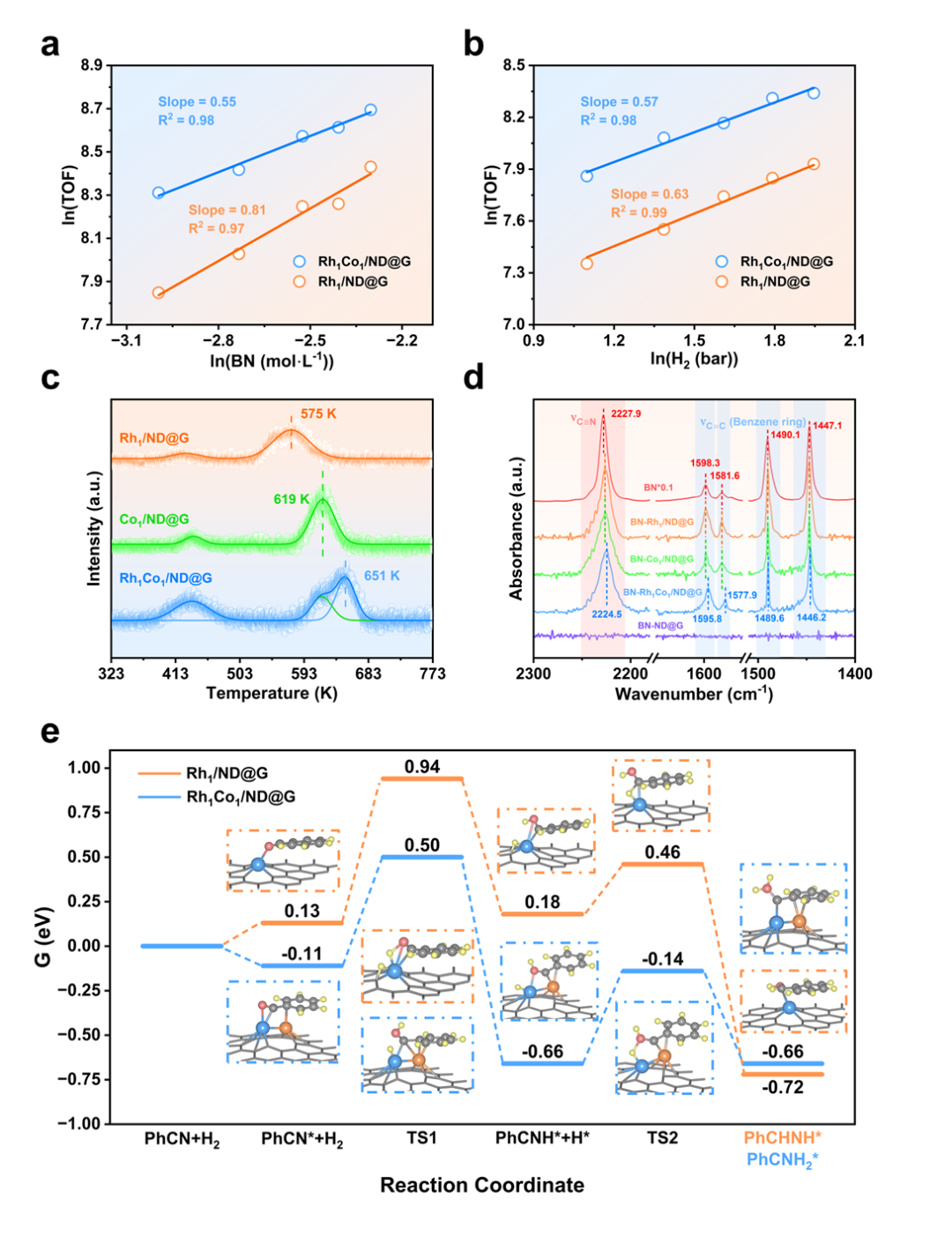
图3. 机理研究。(a-b)动力学研究,(c)BN-TPD,(c)BN-原位红外光谱,(e)DFT计算。

图4. Rh1Co1/ND@G催化BN加氢反应示意图。





