 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库达摩院Lingshu-Cell:离散扩散架构驱动的细胞世界模型
将 ScienceAI 设为星标
第一时间掌握
新鲜的 AI for Science 资讯

在计算生命科学领域,构建 “虚拟细胞” 以模拟生物细胞系统对外部干预的反应,一直是核心科研问题。通过构建高精度的计算模型,研究人员能够在数字空间内预测药物或基因干预后的细胞转录特征,从而极大加速疾病机制研究与药物筛选的进程。尽管现有的单细胞基础模型在学习静态细胞表征方面表现优异,但在精准刻画细胞状态的概率分布以及模拟受扰动后的动态演化过程上,仍面临较大挑战。
为突破这一瓶颈,阿里巴巴达摩院构建了生成式细胞世界模型 Lingshu-Cell。该模型采用掩码离散扩散架构,不仅能够还原全转录组尺度的细胞状态分布,还能精准预测细胞在受到基因扰动或细胞因子刺激后的表达谱响应。这一研究成果标志着单细胞建模从传统的静态描述向生成式模拟迈出了关键一步。

论文地址:https://arxiv.org/abs/2603.25240v1
项目地址:https://alibaba-damo-academy.github.io/lingshu-cell-homepage/
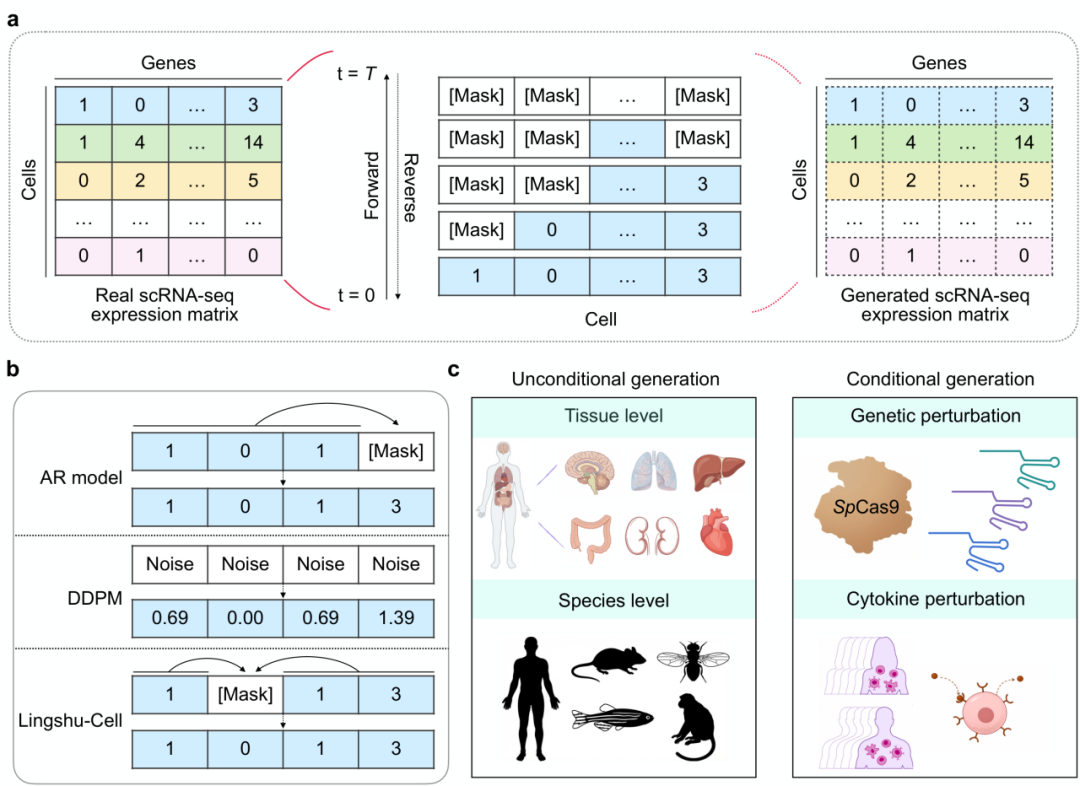
图 1:Lingshu-Cell 总览。(a)从真实 scRNA-seq 表达矩阵到生成表达矩阵的掩码离散扩散模型(MDDM)流程;(b)三种生成范式对比:AR、DDPM、MDDM;(c)应用场景:细胞状态生成(跨组织 / 跨物种)与扰动响应模拟(基因扰动 / 细胞因子刺激)。
虚拟细胞:从数据快照到可预测的世界模型
过去十年,大规模单细胞 RNA 测序(scRNA-seq)技术极大地丰富了人类对不同组织、物种及生理状态下细胞组成与功能的认知。然而,目前大多数分析仍停留在描述性层面,如细胞聚类、注释和静态特征比较,缺乏真正的预测能力。虚拟细胞(Virtual Cells)的愿景正是要改变这一现状:通过构建计算框架,让研究者能够在数字空间中进行大规模实验,进而解析疾病机制并筛选治疗方案。
为了实现这一目标,达摩院团队前瞻性地提出了细胞世界模型(Cellular World Model)概念。类比人工智能领域中能够理解环境规律并支持模拟预测的世界模型,细胞世界模型旨在学习转录组状态的深层表征及其动态演化规律。其核心能力包含两个维度:
状态分布建模:能够捕捉并生成具有生物学意义的细胞异质性,还原真实细胞群体的状态空间分布,而非仅仅学习一个 “平均特征”。
扰动响应预测:能够模拟细胞在特定干预(如基因敲除或信号分子刺激)下的动态演化,预判表达谱的连锁反应。
基于这两个维度的能力,细胞世界模型有望为单细胞研究提供一个高效率、低成本的数字实验预测平台。在这一框架下,研究者不仅可以预演基因扰动后的细胞变化,辅助基因功能研究;还能够比较不同候选药物可能引发的转录组效应,加速药物筛选;并进一步结合个体的细胞信息,对不同治疗方案的潜在效果进行评估。
目前的单细胞基础模型大多局限于学习静态表征,更适合作为下游分析的特征提取器,而非生成式模拟器。为突破此局限,Lingshu-Cell 创新性地引入了当前大模型领域前沿的掩码离散扩散(MDDM)架构。这一架构带来了双重优势:它不仅具备对原始转录组状态分布的强大建模能力,能够精准捕捉高维特征空间中的细胞异质性;更能将细胞状态空间与外部干预条件深度融合,实现了全转录组尺度的精准条件预测。
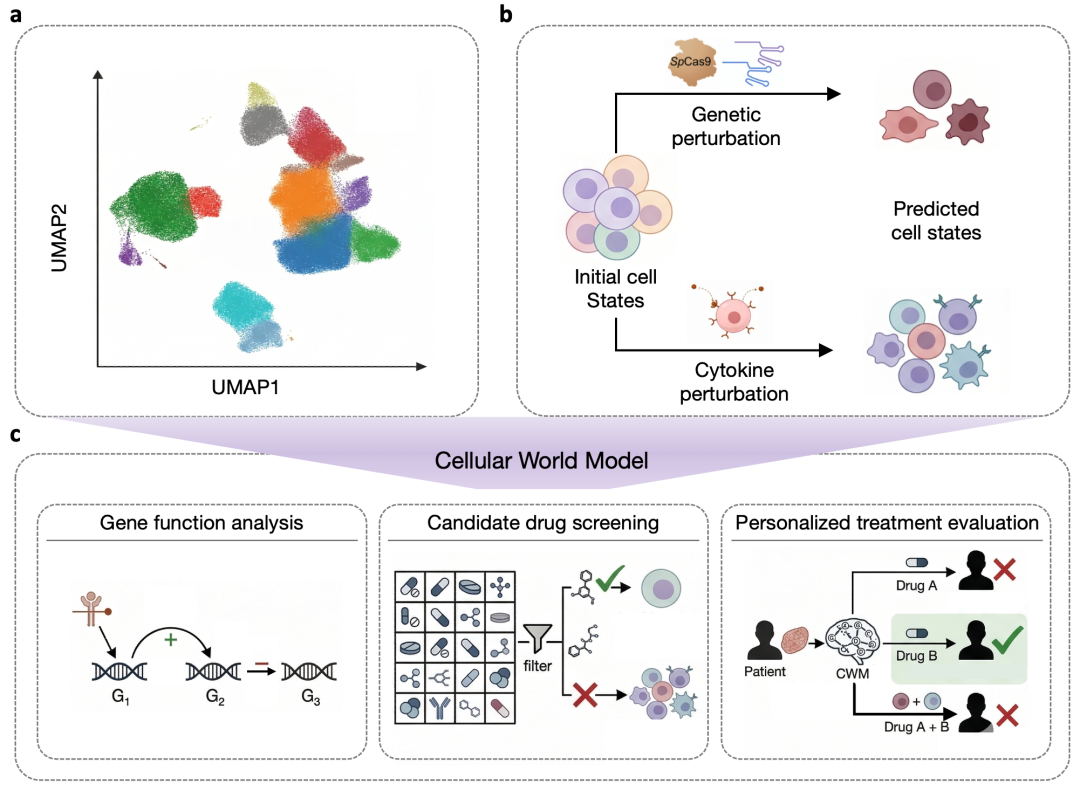
图 2:细胞世界模型核心能力的两个维度,以及潜在应用场景。(a)状态分布建模;(b)扰动响应预测;(c)潜在应用场景,包括基因功能研究、候选药物筛选和个性化治疗评估。
Lingshu-Cell:用离散扩散框架统一建模细胞状态与扰动响应
单细胞转录组数据本质上表现为离散计数数据,而细胞类型、扰动策略等条件信息同样具有离散性。针对这一数据特点, Lingshu-Cell 在架构设计上实现了两项关键创新:
1. 掩码离散扩散模型(MDDM):统一建模的优越性
实现细胞状态分布建模与扰动响应预测相统一的关键是在于将不同的条件与数据纳入同一生成框架下。为此,Lingshu-Cell 采用了大语言模型领域前沿的掩码离散扩散框架进行构建,该框架在处理单细胞数据时展现出以下显著优势:
统一 Token 空间与建模范式:Lingshu-Cell 将细胞类型、外部扰动策略以及基因表达数值等信息,统一转化为离散 Token 并映射至同一个向量空间。这种设计使得模型能够在单一生成范式下,同步完成对 “原始分布” 和对 “各种外部条件响应” 的建模。Lingshu-Cell 无需针对扰动任务设计独立的预测头,而是将扰动视为一种可生成的上下文逻辑,实现在统一范式下的高效训练和泛化。
天然适配数据的离散性:传统 DDPM 使用连续高斯噪声,难以直接应用于单细胞转录组的离散计数数据。Lingshu-Cell 直接在离散 Token 空间操作,天然契合此类生物数据的内在特性。
消除自回归模型的顺序偏置:依托双向 Transformer 架构,Lingshu-Cell 允许每个基因在生成时全局参考全基因组的上下文信息,有效捕捉复杂的协同调控逻辑,避免了单向自回归模型在处理 “无序” 基因序列时产生的人为顺序偏置。
2. 针对全转录组精准建模的技术优化
同时,为了更好地适配全转录组数据的超大规模建模,Lingshu-Cell 引入了多项优化技术,在计算效率与微弱信号捕捉能力之间取得了完美平衡:
多尺度量化编码:针对 UMI 计数跨度大且分布极其倾斜的特点,模型引入了共享量化函数,将原始计数映射到有限的离散状态池中。这一设计在大幅压缩状态空间的同时,通过自适应步长精准保留了低表达量区间的高分辨率信息。
嵌入空间序列压缩:使用 Transformer 直接处理高达 1.8 万个基因的长序列将带来难以承受的计算开销。为此,Lingshu-Cell 引入了嵌入空间的压缩模块,通过随机分组(Random Grouping)和线性投影,将超长基因序列压缩为高信息密度的表征(如将长度从 18,000 维大幅压缩至 500-2,000 维)。这不仅成倍降低了计算成本,更通过多基因信号的线性混合有效过滤了单基因层面的噪声干扰。
条件引导与生物学先验注入:为了让模型在上万个基因中精准定位扰动方向,Lingshu-Cell 引入了条件引导(CFG)机制。通过在采样阶段强化目标条件信号,模型能够更明确地向指定的扰动状态演化。同时,模型支持注入已知生物学先验,通过在采样起点锚定关键基因状态,引导生成结果严格符合真实生物学规律。
通过统一建模的掩码离散扩散架构与针对全转录组的技术优化,Lingshu-Cell 在适配单细胞数据特性的同时,有效解决了超长基因序列建模中计算效率与精度难以兼顾的难题。这种设计与工程的协同,让模型得以精准刻画单细胞转录组的真实分布规律,进而生成高保真、条件可控的单细胞基因表达谱。
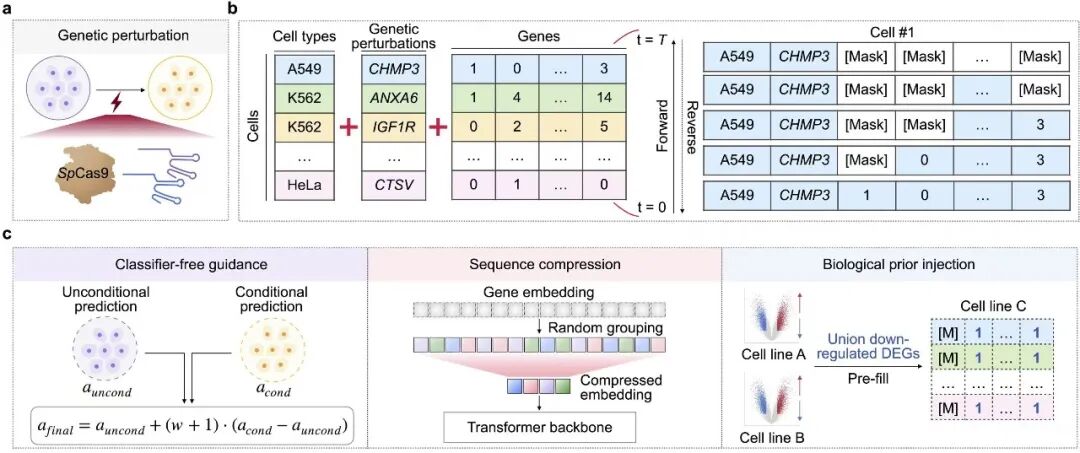
图 3:以基因扰动为例,展示模型框架与关键设计。(a)基因扰动任务示意;(b)输入序列构成与掩码离散扩散过程;(c)三个关键技术:Classifier-free guidance、序列压缩、生物学先验注入。
实验验证:还原异质性,完成状态分布建模任务
1. 跨组织与跨物种的强大建模能力
在覆盖 9 个人类组织(脑、心脏、肺、结肠等)和 4 个非人物种(小鼠、恒河猴、斑马鱼、果蝇)的单细胞转录组数据上,Lingshu-Cell 展现出了卓越的泛化与细胞状态分布建模能力。为了严谨验证这一能力,研究团队主要从三个维度展开了系统评估:(1)整体分布是否匹配,(2)各细胞亚型的组成比例是否一致,(3)经典 marker 基因的表达模式是否被精准复刻。
在 PBMC 数据上,生成细胞与真实细胞在 UMAP 中呈现出高度一致的群体结构;各亚型的组成比例也被较好重现,有效避免了传统生成模型中常见的模式崩溃(如过度生成或遗漏特定亚型)问题;marker 基因的空间表达模式同样与真实数据吻合。这说明模型学到的绝不是浅层统计学的相似性,而是具有生物学意义的细胞状态结构。在定量比较中,Lingshu-Cell 在 5 个评估指标上均优于现有基准模型:scDiffusion 和 scVI。这种优异的表现也不仅限于单一数据集:从人类脑组织到果蝇,不同组织和不同物种的数据上都得到了稳定且一致的验证。
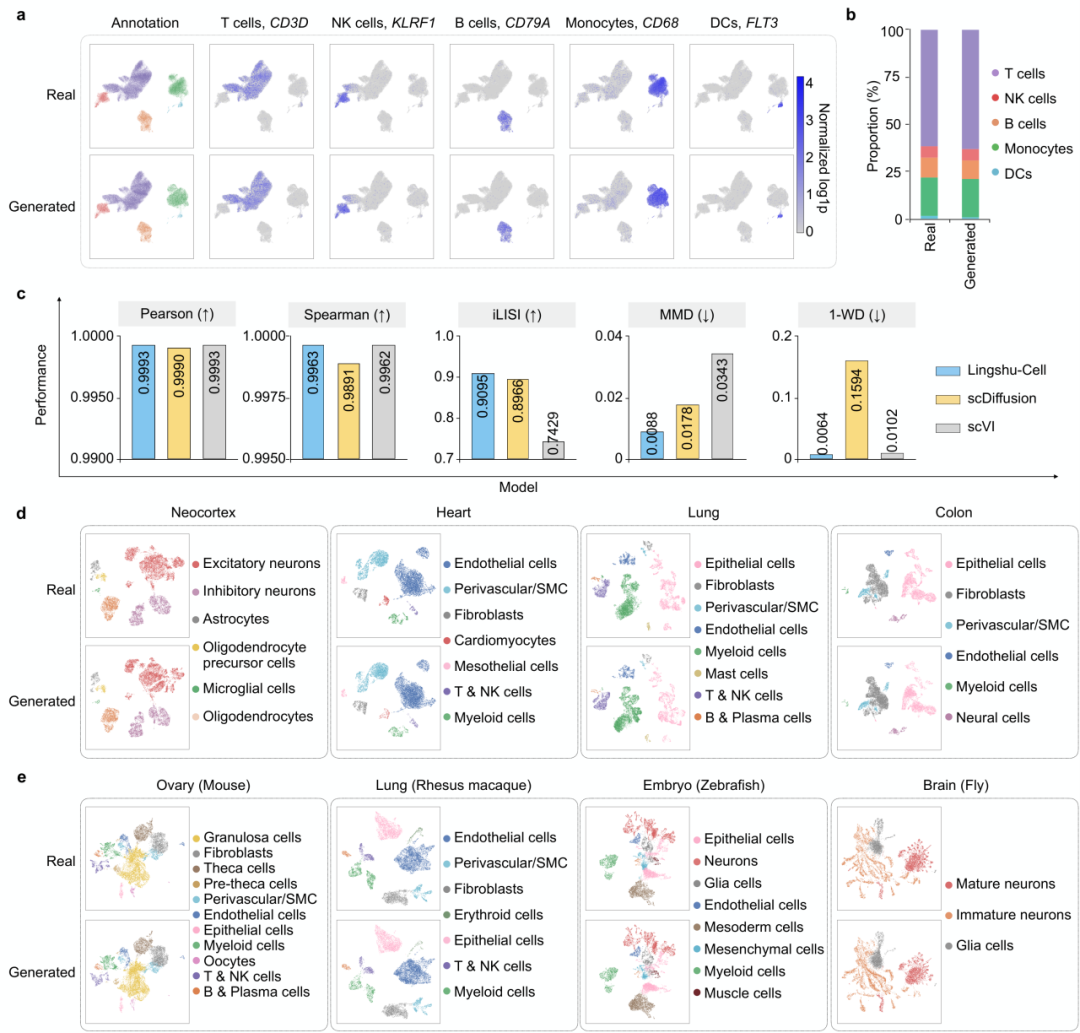
图 4:细胞状态生成结果。(a)免疫细胞 UMAP 可视化与 marker 基因表达对比;(b)细胞亚型组成比例;(c)定量 benchmark(vs scDiffusion, scVI);(d-e)跨人类组织与跨物种的嵌入结构对照。
2. 从主要谱系到细粒度亚型的高分辨率刻画
更进一步地,在更高分辨率的分析下,Lingshu-Cell 生成的细胞同样能够精细刻画真实数据中的细胞亚型结构。无论是 UMAP 中更细粒度的亚型分布,还是不同细胞亚型的组成比例,生成数据均与真实数据保持了较高保真度。从主要细胞谱系到更细粒度的细胞亚型,Lingshu-Cell 都能较好重现真实数据中的分布特征,标志着该模型已经能够对复杂细胞状态分布进行全景建模。
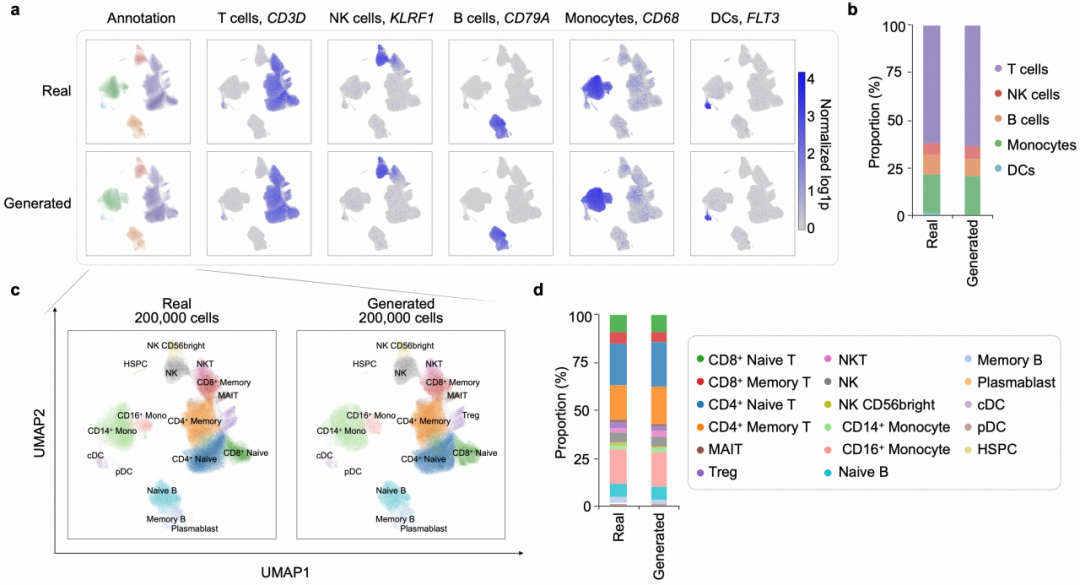
图 5:更大规模细胞群体中的高分辨率细胞状态生成结果。(a)PARSE 10M PBMC 数据中真实细胞与生成细胞的 UMAP 可视化,并按细胞类型注释与 canonical marker 基因表达着色;(b)真实数据与生成数据中主要细胞类型比例对比;(c)在更高分辨率下的 UMAP 可视化,显示生成细胞能够重现真实数据中的细胞亚型结构;(d)真实数据与生成数据中细胞亚型比例对比,显示在更高分辨率下仍保持稳健一致性。
应用场景:高保真推演,实现多扰动条件响应预测
在真实的生物学研究中,科学家们往往通过施加外部干预(如敲低 / 敲除某个特定基因、使用特定药物或施加细胞因子刺激)来观察细胞的反应,进而探究疾病机制或验证药物靶点。扰动响应预测,即是利用计算模型在数字空间中预演这一生物学过程。这一任务的目标是预测细胞在受到特定刺激后,成千上万个基因表达谱的动态级联变化,这正是细胞世界模型的核心能力之一。Lingshu-Cell 在两大扰动条件上进行了实验,分别是基因敲除扰动和细胞因子扰动。
1. 基因扰动响应预测:基因扰动是指通过 CRISPR 等基因编辑技术,特异性地抑制或敲除目标基因的表达,进而观察细胞在该基因受扰动后成千上万个基因的转录表达级联变化,这也是去年虚拟细胞竞赛 (Virtual Cell Challenge) 的核心任务。 VCC 由 Arc Institute 发起、NVIDIA 与 10x Genomics 赞助,是单细胞基因扰动响应预测领域的全球性比赛。我们将 Lingshu-Cell 与 Generalist Rank(综合考核 7 项核心指标)的当前榜单队伍进行了全面对比。Lingshu-Cell 在这个榜单上取得了最优的平均排名,尤其是在 MAE(平均绝对误差)和 Pearson-Δ 两项关键指标上的表现排名第一。这不仅印证了模型卓越的泛化预测能力,更验证了 MDDM 架构对于基因扰动响应预测任务上的巨大潜力。
2. 细胞因子扰动响应预测:在更大规模的 PARSE 10M PBMC 数据集上(涵盖 12 个供体 × 90 种细胞因子),Lingshu-Cell 同样在与多个基准模型的对比中取得了最优的综合评分。该任务的核心挑战在于,同一种刺激在不同供体背景下可能引发不同的转录组响应,而不同刺激之间的效应也并不相同。模型需要捕捉这种条件依赖的差异,而不是简单学习一种平均模式。
基因扰动和细胞因子刺激代表了两类底层机制截然不同的生物学干预,而 Lingshu-Cell 在这两类任务上都取得了领先,有力证明了这一统一框架不仅能够精确建模细胞状态分布,更能够进一步模拟不同类型干预所引发的复杂细胞变化。
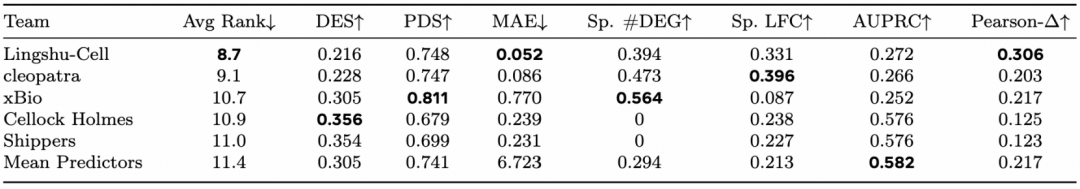
表 1:VCC H1 benchmark 赛后评测综合排名。
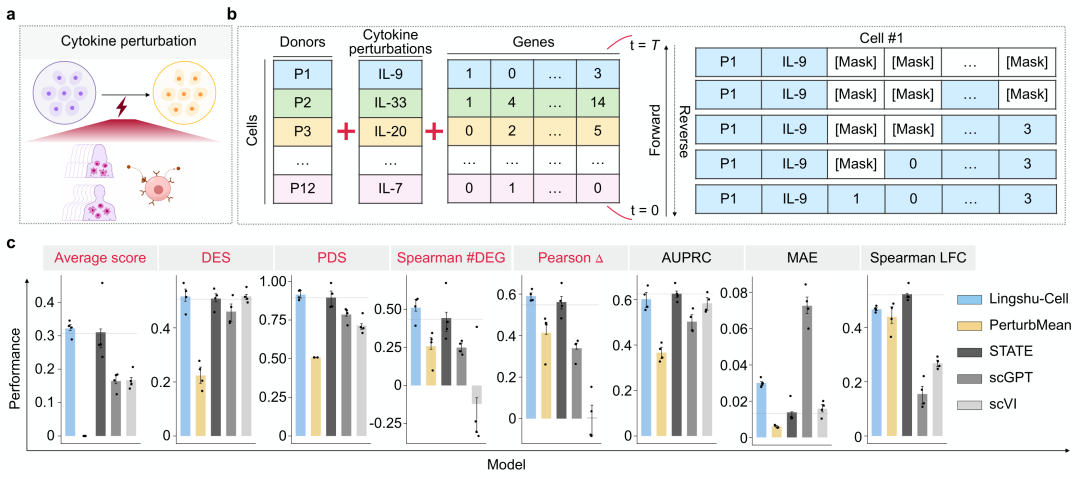
图 6:PARSE 细胞因子扰动响应预测的多指标对比。(a)细胞因子诱导转录组扰动的示意图;(b)输入序列构成与掩码离散扩散过程;(c)Lingshu-Cell 与多个基准模型在 PARSE 10M PBMC 数据集上的性能对比。评估涵盖 1 项综合评分(Average score)与 7 项核心指标。红色标注代表 Lingshu-Cell 在该指标上超过其他所有对比的基准模型。
虚拟细胞还有多远?
在这篇工作中,我们介绍了 Lingshu-Cell,一个采用统一离散扩散架构实现细胞状态建模和扰动响应的预测的细胞世界模型。尽管目前的探索仍局限于转录组层面,且数据层面的高保真模拟尚未完全揭示细胞演化的底层因果机制,距离构建真正意义上的 “虚拟细胞” 仍有显著差距。但作为通向细胞世界模型的一次初步尝试,Lingshu-Cell 充分证实了离散扩散范式在单细胞生成建模领域的巨大潜力。展望未来,随着表观遗传、蛋白质组学等多模态数据的逐步引入,计算生命科学从静态分析向动态模拟演进的趋势将越来越清晰。整个行业正加速向全面动态数字模拟的新阶段大步迈进,一个属于数字生命的时代已然拉开序幕。
人工智能 × [ 生物 神经科学 数学 物理 化学 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。





