 五度妙笔
五度妙笔 企业透视镜
企业透视镜 API商城
API商城
 数据库
数据库820万合成分子、100%化学有效:CoCoGraph用534K级参数把分子生成推向真实化学分布
将 ScienceAI 设为星标
第一时间掌握
新鲜的 AI for Science 资讯


编辑丨&
发现与开发新分子效能是现代化学的重要研究事业之一。从新药的开发到更可持续材料的创造,一切都依赖于寻找具有有用性质的新原子组合。分子生成之所以难,不只是因为「要新」,而是因为化学空间太大、规则太硬。
2026 年 5 月 4 日,Universitat Rovira i Virgili(URV)的团队提出了 CoCoGraph——一个协作式、受约束的离散图扩散模型,目标不是只「生成像分子」的图,而是生成保证化学有效、同时又尽可能接近真实分布的分子。
在这篇发表于《Nature Machine Intelligence》上的「A collaborative constrained graph diffusion model for the generation of realistic synthetic molecules」中,CoCoGraph 不仅实现了 100% 的化学有效性,还把模型参数量砍到了同类顶尖水平的 1/10。

论文链接:https://www.nature.com/articles/s42256-026-01229-5
双边交换约束机制
在 CoCoGraph 出现之前,主流的图扩散模型(如 DiGress)虽然能生成分子,但存在两个致命伤。
AI 可能会给碳原子连上 5 个键,或者让氢原子孤悬。这些在数学上成立但在化学上「非法」的结构,极大地浪费了后续筛选的算力;而模型为了捕捉分子内部复杂的原子关联,往往需要数百万甚至上千万的参数,这让训练和推理变得异常沉重。
所以 CoCoGraph 的思路非常硬核。与其在生成后去剔除错误的分子,不如在生成的每一步都加入物理约束,让模型只能生成正确的分子。
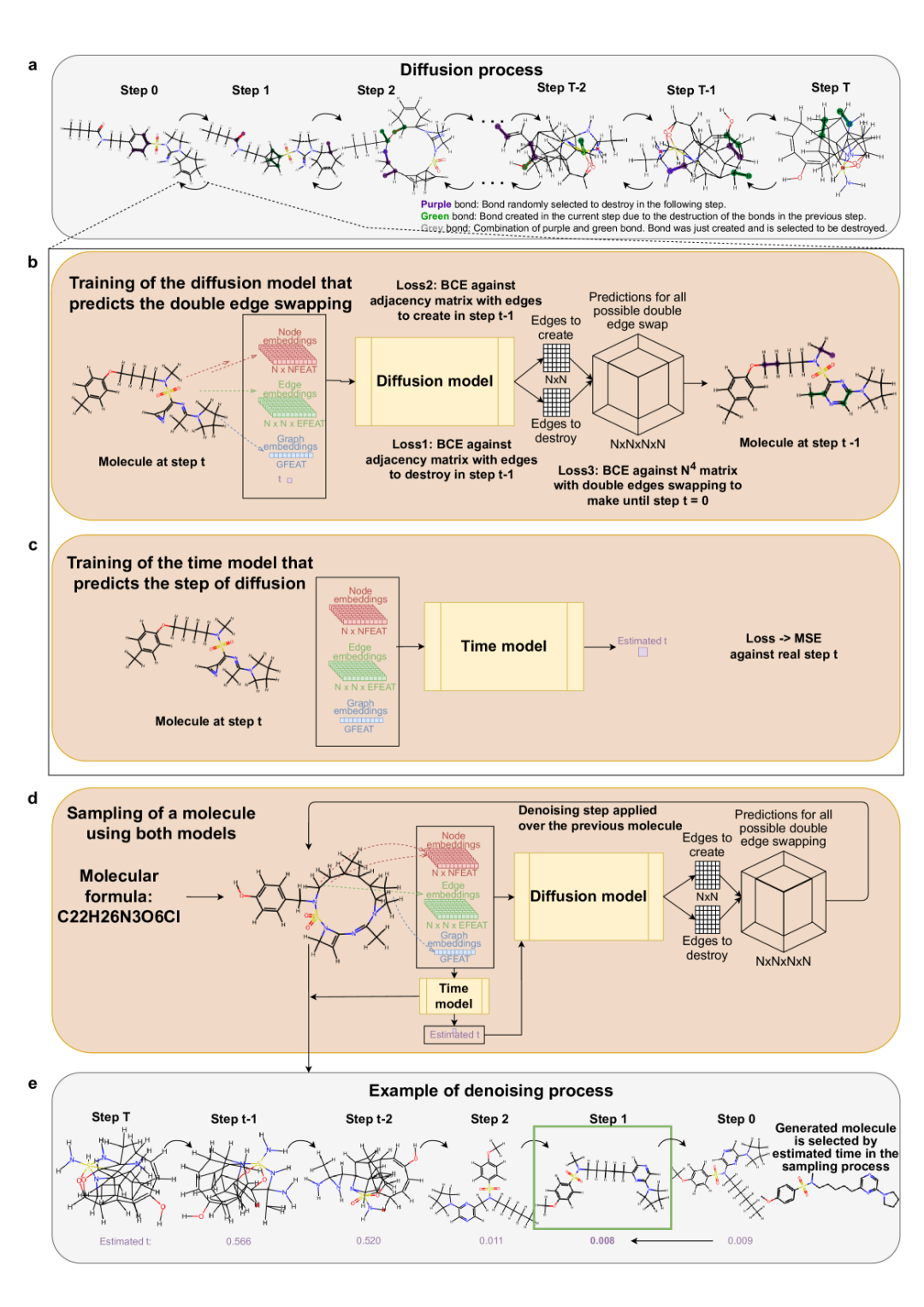
图 1:受限协作图扩散模型,CoCoGraph。
这就是 CoCoGraph 的核心设计:把分子扩散过程定义成一种离散的「双边交换」(double edge swapping, DES)操作。每一步不再随意加噪声,而是随机选取两条化学键并删除,再重连成两条新键,从而在 noising 和 denoising 的每个阶段都保持原子数、分子式和度序列,也就是价态约束。
作者强调,这样做的结果是,凡是不满足化学约束的图根本不会进入生成空间,模型也不需要学会化学规则本身,只需要学习真实分子更细微的结构模式。为了让这个过程既稳又小,BASE 版只需要约 534K 参数,远少于对比模型 JTVAE 的 5.3M 和 DiGress 的 4.6M。
对于生成过程,特殊的协作机制发挥了它的作用。扩散模型预测下一步应撤销哪一组双边交换,时间模型则估计当前图离原始分子还有多远。前者接收分子图特征和扩散时刻,输出所有可能 DES 的概率;后者输入当前分子图,回归一个 0 到 1 之间的归一化时间值。
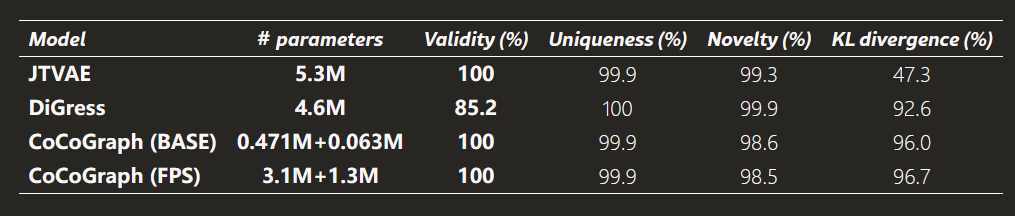
图 2:基于GuacaMol基准的模型对比。
得益于这两种设计,BASE 版和 FPS 版都达到了 100% chemical validity,而且没有牺牲多样性:两者的 uniqueness 都是 99.9%,novelty 分别为 98.6% 和 98.5%。
更真实的化学,而不只是某类化学
作者随后把评价尺度扩展到 36 个化学性质,涵盖分子大小、拓扑、电子性质和药物相似性等指标。结果显示,CoCoGraph 在 36 项里有 23 项优于 DiGress,33 项优于 JTVAE;在选出的 10 个代表性属性中,它对 heavy atom count、valence electrons 等多项性质的分布拟合都更接近原始分子。
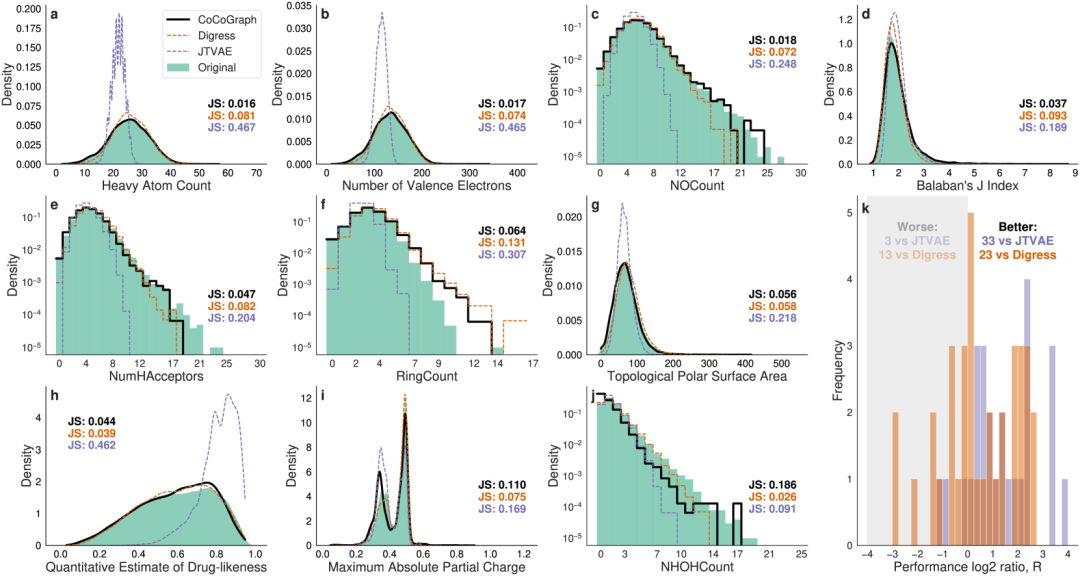
图 3:对 36 种化学性质子集进行详细性能比较。
此外,因为模型足够轻,CoCoGraph 能在单块中端 GPU 上每小时生成成千上万条有效分子。团队据此构建了一个包含 8.2 million 条合成分子的数据库,其中只有 7.1% 是冗余,换言之大约 7.6 million 条是新颖且有效的分子结构。
为了验证这些结构是否真的「像化学」,团队还做了一个分子版图灵测试:把真实分子和 CoCoGraph 生成分子配成同分子式的对照对,交给 102 位有机化学相关背景的参与者判断。总共 2040 次判别中,专家平均准确率只有 62%;本科背景者是 59%,研究生背景者是 64%。
更微妙的是,对于无环分子和以脂肪族键为主的分子,参与者表现甚至接近 50% 的随机猜测水平,说明这类生成结果已经相当接近真实分子的视觉与结构直觉。
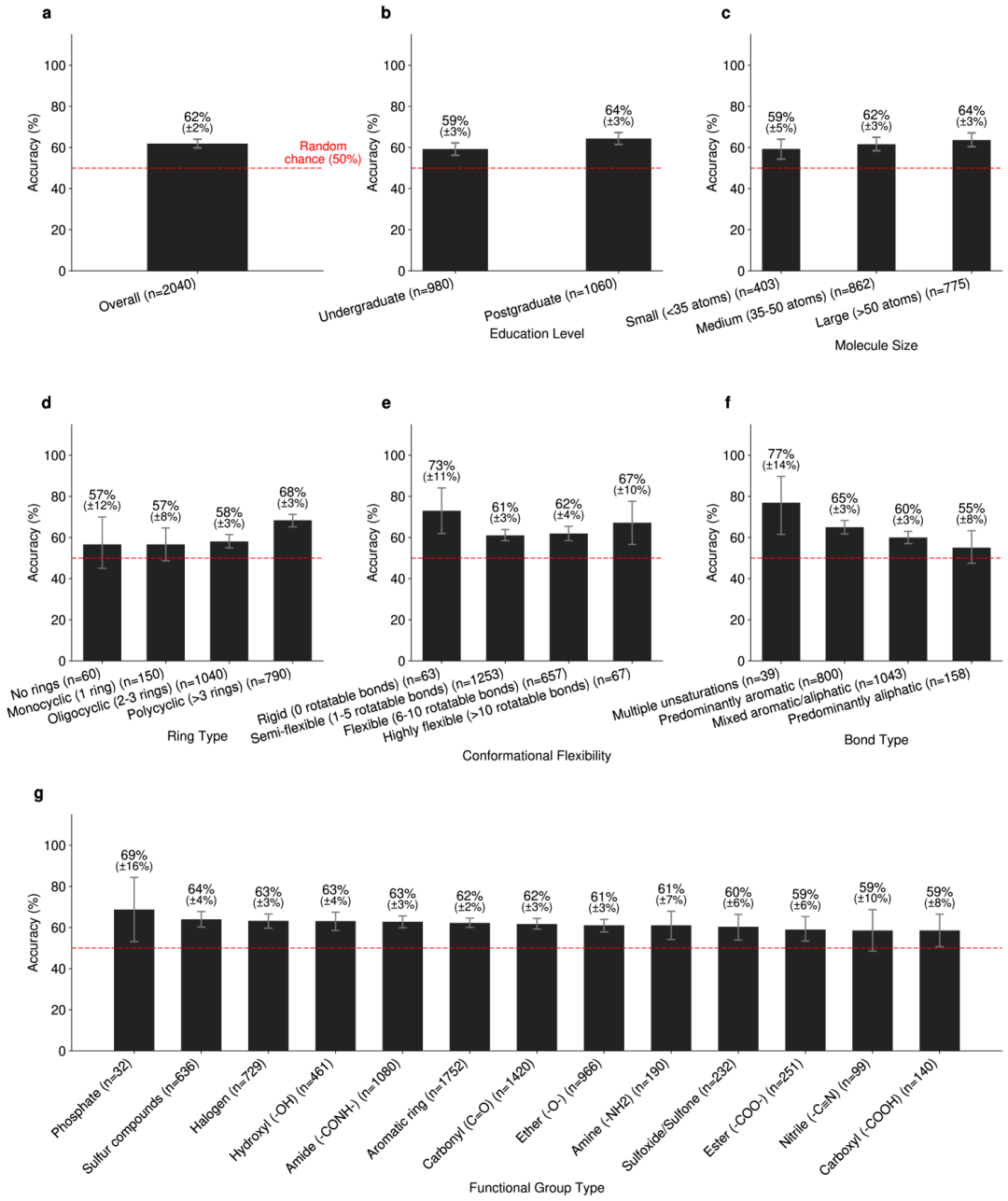
图 4:在图灵类测试中表现。
挑战真实分子生成
研究团队明确表示这只是开始。中期到长期的主要目标是能够向人工智能请求具有特定性质的分子;例如,请求一种可溶性、无毒且适用于特定应用的分子。他们将目标锁定在分子生成上,并期望着能将特定目标应用于这一过程。
对药物开发、材料设计、催化和环境分子发现来说,这意味着分子生成不必先在无效空间里空转,而可以直接在可行化学空间里做高密度探索。该研究给出的 8.2 million 分子数据库,也因此不只是一个结果展示,而是一张能继续被下游任务调用的化学地形图。
相关报道:https://phys.org/news/2026-05-chemistry-aware-ai-generate-millions.html
人工智能 × [ 生物 神经科学 数学 物理 化学 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。





