“「药企合规」系列第二篇。第一篇讲合规体系由什么构成,第三篇从产品经理视角谈怎么把合规做进产品里。最后会聊下 AI 拜访 Agent 的成败, Pre-call 和 Post-call 的合规问题。
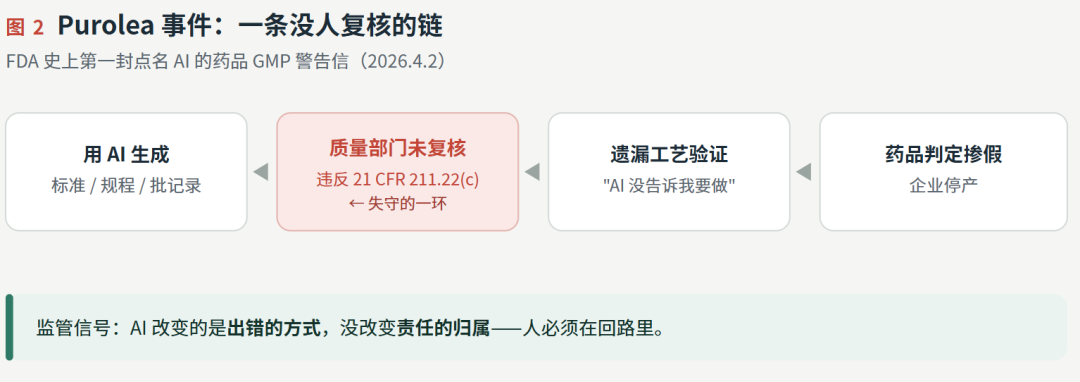
2025 年 10 月底,FDA 的检查员走进密歇根州利沃尼亚一家叫 Purolea 的小厂。它名字里带"Cosmetics",实际生产顺势疗法的非处方药,所以归 21 CFR 210/211 这套药品 GMP 管。检查中查出:这家厂没做过工艺验证就把药放了出去。厂主的解释是——她不知道要做工艺验证,因为"AI 没告诉她"。她用 AI 智能体生成了药品标准、操作规程和批生产记录,没人复核,直接拿去用了。
2026 年 4 月 2 日,FDA 据此发出警告信(编号 320-26-58)。这是 FDA 历史上第一封专门点名"不当使用人工智能"的药品 GMP 警告信。
把这件事讲清楚,就讲清楚了 2026 年 AI 药企合规的全部要害。这一年,中美欧三大监管体系几乎同时出牌,看起来各管各的,底层却是同一句话:监管不反对你用 AI,监管反对的是没有人对 AI 的产出负责。 谁负责、怎么留痕、哪些环节 AI 根本不能碰——这三问,就是这篇要拆的三张底牌。
一、美国:从"原则共识"到"开第一张罚单"
美国这一年走完了"立原则—落执法"两步。
第一步,2026 年 1 月 14 日,FDA 与 EMA(欧洲药品管理局)罕见地联合发布《药物开发中良好 AI 实践的指导原则》,一口气给出 10 条原则。它不是强制法规,而是把双方对 AI 的监管预期摆上桌:以人为本、风险分级、明确使用场景(COU,即这个 AI 到底用在哪、解决什么问题,必须事先界定清楚)、数据治理、全生命周期监控、信息透明。核心是一个词——可信度:你得拿出证据证明,这个模型在它声称的场景里靠得住,而且会因数据漂移(环境变了、模型表现悄悄退化)而被持续监测。
第二步,就是开头那封 Purolea 警告信。这里要纠正一个流行的误读:它不是 FDA 在排斥 AI。恰恰相反,FDA 自己 2025 年 6 月就上线了内部 AI 工具 Elsa,2026 年还在迭代。FDA 罚的不是"用了 AI",而是"用了 AI 却没人复核"——违反的是 21 CFR 211.22(c),即质量部门必须对所有规程和标准负审批责任。律师对此案的总结很直白:问题不在 AI 起草文件,而在没有任何懂 GMP 的人去看一眼。
放进大背景看:FDA 在 2025 财年发了 303 封药品警告信,比上一年涨了约六成。执法本就在收紧,Purolea 只是第一个把"AI"三个字写进违规栏的。
二、欧盟:把"哪些 AI 不能碰关键环节"写进 GMP
如果说美国靠个案立规矩,欧盟则在直接改 GMP 条文。
欧盟正在为 EudraLex 第四卷新增附件 22《人工智能》,与修订版附件 11(计算机化系统)一起,构成一次"数字化 GMP 大修"。附件 22 草案(2025 年 7 月版)划了一条很硬的线:动态、自适应、概率性的模型——也就是生成式 AI 和大语言模型(LLM)——不得用于关键 GMP 环节,只能用在文档起草、培训这类非关键场景,且必须有人工监督和适用性核查。它要求 AI 模型记录特征归因、给出置信度、保留人在回路(HITL)的复核节点。
(这条"GenAI/LLM 不得碰关键 GMP",是欧盟的规则,不是 FDA 的——后面对比表会再点一次,因为这是最容易张冠李戴的地方。)
EMA 将于 2026 年 6 月 30 日至 7 月 1 日召开专家研讨会,为附件 22 定稿收集意见——第一天开放陈述,第二天闭门讨论。也就是说,这条线还没最终落地,正在拍板的路上。
至于常被一起提的欧盟《AI 法案》,要特别小心日期:2026 年 8 月 2 日生效的,是法案主体、透明度义务和执法机制的启动;而最受关注的高风险义务已被推迟——2026 年 5 月达成的"数字综合法案"(Digital Omnibus)政治协议,把独立高风险系统(附件 III)推到 2027 年 12 月,把嵌入医疗器械等受监管产品的 AI(附件 I)推到 2028 年 8 月(正式文本仍在走通过程序)。换句话说,"药物/器械相关 AI 的高风险合规大限就在 2026 年 8 月"这种说法,已经过时。
三、中国:监管自己先用上 AI,再要求产业跟上
中国的打法又不一样——监管侧先智能化,倒逼产业侧数智化。
2026 年 4 月 2 日,国家药监局发布《关于"人工智能+药品监管"的实施意见》(国药监综〔2026〕6 号),是"人工智能+"行动在药监领域的首份纲领性文件,覆盖药品、医疗器械、化妆品("两品一械")全生命周期。它提出七大监管场景的智能化(智能审评审批、智慧检查、检验监测、政务服务等),定下两个时间点:2030 年初步建成融合体系,2035 年形成数智驱动的治理新格局。
最该被企业记住的是那条贯穿始终的红线——"数智赋能、人工复核、全程留痕"。和大洋彼岸的 Purolea 一个意思:AI 可以提效,但人必须在回路里,过程必须可追溯。文件同时明确 AI 在审评审批中是辅助定位,不得替代人工决策;并点名血液制品、中药注射剂等高风险品种要"生产、检验全程数智化"。
四、三家说的,其实是同一件事
把中美欧摆到一张表上,分歧主要在形式(强制法规 / 原则 / 纲领),共识却高度一致。
剥到底,三家压在同一根"合规脊柱"上:
- 人在回路:AI 起草、AI 识别、AI 打分都行,但最终对产出负责的必须是人。
- 风险分级:越靠近关键环节(放行、诊断、批记录),约束越严,GenAI/LLM 甚至被欧盟挡在门外。
- 全程留痕 / 可解释:谁在什么时候用 AI 生成了什么、谁复核的,要查得到;黑箱不被接受。
- 数据治理:训练数据质量、用途、出境,都要管。
这根脊柱,就是第三篇要回答的产品问题:怎么把它做进产品里,而不是事后补。
对有 SaaS 交付 + 医疗场景背景的人,下面这几条边界尤其要盯(图 5):
- HCP(医生)数据:用于 AI 训练或商业分析,必须有明确授权或合法来源(《个人信息保护法》),不能靠灰色手段抓取。
- AI 诊疗建议:一旦给出诊断或用药推荐,很可能落入"软件作为医疗器械"(SaMD,即软件本身被当作医疗器械监管),触发《医疗器械监督管理条例》的审批——这条对做器械、做诊断的人最致命。
- 电子处方 / DTP 流转:符合《处方管理办法》,接入医保监管平台。
- 药物警戒里的 AI:用 AI 做信号检测可以,但要保留可解释性和人工复核节点。
- 跨境数据:涉及人类遗传资源、数据出境的,先过安全评估。
这一年,规则从"讨论"变成了"执法"
回头看 2026 这半年,AI 药企合规走完了三级跳:从 1 月的 FDA-EMA 原则共识,到 4 月 Purolea 的第一张罚单,再到中国"两品一械"全链条的纲领部署——监管的语气从"我们在研究"变成了"我们开始罚"。
往前看,三件事大概率发生:可信度证据会像稳定性数据一样,成为新一类必交的申报材料;人在回路会从一句口号,变成一个可被审计的、有时间戳和签名的具体节点;而中国"监管先用 AI、再拉产业"的模式,可能跑出一条和欧美都不同的路径——监管自己的智能体,反过来定义了企业该长什么样。
Purolea 那位厂主把锅甩给 AI,FDA 没接。这恰恰说明:AI 改变的是出错的方式,没改变责任的归属。下一篇,我们就从产品经理的视角,聊聊怎么把"人在回路、风险分级、全程留痕"这根脊柱,从合规要求变成产品的出厂设置。我愿意和读者一起期待。
写作附注
自我批评:这篇最弱的是欧盟那节。AI 法案的推迟还在走正式通过程序、附件 22 也还没定稿,等于我在写"正在变的东西",三个月后可能就要更新——读者拿到时务必看一眼最新进展。
建设性追问:如果三家监管的共识真的是"人在回路 + 风险分级 + 全程留痕",那对产品经理来说,真正的问题就变成——这三样到底该做成产品里用户能看见的功能,还是用户感觉不到的底层约束?做错了方向,要么没人用,要么不合规。这是第三篇的核心。
名词速查表
- COU(Context of Use):使用场景,指一个 AI 模型具体用在哪、解决什么问题,必须事先界定。
- 可信度(Credibility):用证据证明模型在其声称场景里靠得住的程度。
- 数据漂移(Data Drift):环境变化导致模型表现随时间悄悄退化。
- 21 CFR 211.22(c):美国 GMP 条款,质量部门须对规程和标准负审批责任。
- 附件 22(Annex 22):欧盟 GMP 新增的 AI 专章(草案),限制 GenAI/LLM 用于关键环节。
- 人在回路(HITL):AI 流程中保留人工复核与最终决策的节点。
- SaMD:软件作为医疗器械,软件本身被当作医疗器械监管。
- PV(药物警戒):药品上市后不良反应监测与风险管理。
 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库