 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库FDA警告信:鉴别检测不充分、设备清洁和维护不足等


WARNING LETTER
警告信
美国FDA于2025年12月对美国Sklar Personal Care Inc公司出具一封警告信。信中指出该公司被审查出多项违反CGMP的情况,包括鉴别检测不充分、设备清洁和维护不足、工艺控制程序不足、未建立质量控制部职责等方面。缺陷情况汇总如下:
1、该公司未能对药品的每个组分进行至少一项鉴别检测:该公司未对用于生产非处方药品的原料进行鉴别测试,包括未对某原料进行甲醇检测,FDA认为没有充分的测试,就没有科学证据证明原材料在用于药品生产前符合适当的规格。作为生产商,有责任在生产使用前对药品组分进行取样、测试和检查,以确保其质量合格。
2、该公司未能建立并遵循充分的设备清洁和维护书面程序,具体表现为:该公司缺乏公司OTC药品书面程序所要求的清洁验证研究。未能证明清洁操作足以去除潜在污染物,并防止在用于生产该公司OTC产品的共用设备(例如,灌装机和用于配制的小型器具)中出现产品残留。清洁过程中未能充分去除生产设备上的残留物,可能导致后续在非专用设备上生产的药品受到污染。
3、该公司未能建立充分的生产和工艺控制书面程序,以确保生产的药品具有其声称或标明的特性、规格、质量和纯度,具体表现为:未能对所有非处方(OTC)药品进行工艺验证,包括但不限于某产品。
4、该公司未能建立适用于质量控制部门的充分书面职责和程序,且未能遵循此类书面程序,具体表现为:缺乏对OTC药品生产的充分质量部门(QU)监督,未对自2022年以来生产的OTC药品进行年度产品回顾(APR),未能遵循原材料取样程序,因为取样区存放了多种不同的物料,这带来了潜在污染和混淆的风险。

The United States Food and Drug Administration (FDA) inspected your drug manufacturing facility, Sklar Personal Care Inc. (DBA BOV Solutions), FEI 3007953761, located at 1105 E Garner Bagnal Boulevard, Statesville, North Carolina 28677, from May 19 to May 23, 2025 and June 9, 2025.
美国食品药品监督管理局(FDA)于2025年5月19日至5月23日以及2025年6月9日,对位于北卡罗来纳州斯泰茨维尔市加纳·巴格纳尔东大道1105号(邮编28677)的你们公司药品生产工厂Sklar Personal Care Inc.(经营名称为BOV Solutions,FEI 3007953761)进行了检查。
This warning letter summarizes significant violations of Current Good Manufacturing Practice (CGMP) regulations for finished pharmaceuticals. See Title 21, Code of Federal Regulations (CFR), parts 210 and 211 (21 CFR parts 210 and 211).
本警告信概述了成品药品现行药品生产质量管理规范(CGMP)方面的重大违规行为。参见《美国联邦法规》第21篇第210和211部分(21 CFR parts 210 and 211)。
Because your methods, facilities, or controls for manufacturing, processing, packing, or holding do not conform to CGMP, your drug products are adulterated within the meaning of section 501(a)(2)(B) of the Federal Food, Drug, and Cosmetic Act (FD&C Act), 21 U.S.C. 351(a)(2)(B).
由于你们公司用于生产、加工、包装或储存的方法、设施或控制不符合CGMP,根据《联邦食品、药品和化妆品法案》(FD&C Act)第501(a)(2)(B)条(FD&C Act), 21 U.S.C. 351(a)(2)(B)),你们公司的药品被视为掺假产品。
We reviewed your June 16, 2025, response to our Form FDA 483 in detail and acknowledge receipt of your subsequent correspondence.
我们详细审查了你们公司于2025年6月16日对我们FDA 483表的回复,并确认收到你们公司后续的函件。
During our inspection, our investigators observed specific violations, including but not limited to the following:
在我们的检查期间,我们的调查员观察到了具体的违规行为,包括但不限于以下内容:
1. Your firm failed to conduct at least one test to verify the identity of each component of a drug product (21CFR 211.84(d)(1).
你们公司未能对药品的每个组分进行至少一项鉴别检测(21 CFR 211.84(d)(1))。
Your firm failed to adequately test your incoming components for identity before using the components to manufacture your OTC drug products. Specifically, your firm did not perform identity testing for the following raw materials: (b)(4), which are used in the production of (b)(4) batch (b)(4).
你们公司在使用组分生产你们公司的非处方(OTC)药品之前,未能充分对来料组分进行鉴别测试。具体而言,你们公司未对以下用于生产(b)(4)批次(b)(4)的原材料进行鉴别测试:(b)(4)。
We also note that you use raw material (b)(4) to manufacture your OTC drug products. However, testing for the presence of methanol is not performed for incoming (b)(4).
我们还注意到你们公司使用原材料(b)(4)来生产你们的OTC药品。然而,你们未对来料的(b)(4)进行甲醇存在检测。
The use of (b)(4) contaminated with methanol has resulted in various lethal poisoning incidents in (b)(4).
使用受甲醇污染的(b)(4)曾在(b)(4)导致多起致命中毒事件。
Without adequate testing, you do not have scientific evidence that your raw materials conform to appropriate specifications before their use in the manufacture of your drug products. As a manufacturer, you have a responsibility to sample, test, and examine drug components before their use in production to ensure adequate quality.
没有充分的测试,你们就没有科学证据证明你们的原材料在用于药品生产前符合适当的规格。作为生产商,你们有责任在生产使用前对药品组分进行取样、测试和检查,以确保其质量合格。
In your response, you commit to updating testing specifications and prioritizing the identity testing of raw materials on order for the remaining production planned in 2025. You also commit to ensuring that every (b)(4) container delivered will be tested for methanol by July 31, 2025.
在你们的回复中,你们承诺更新检测程序,并优先对2025年计划剩余生产订单所需的原材料进行鉴别测试。你们公司还承诺确保在2025年7月31日前对交付的每个(b)(4)容器进行甲醇检测。
Your response is inadequate. You did not provide a risk assessment addressing the use of untested raw material in your finished OTC products. In your response, you did not commit to testing the remaining raw materials or the retain samples of the OTC drug products already distributed.
你们公司的回复不充分。你们未提供风险评估,以解决在成品OTC产品中使用未经测试原材料的问题。在你们的回复中,你们未承诺对剩余的原材料或已分销OTC药品的留样进行测试。
In response to this letter, provide:
作为对本信的回应,请提供:
A comprehensive review of your material system to determine whether all suppliers of components, containers, and closures are each qualified, and the materials are assigned appropriate expiration or retest dates. The review should also determine whether incoming material controls are adequate to prevent use of unsuitable components, containers, and closures.
对你们的物料系统进行全面审查,以确定所有组分、容器和密封件的供应商是否均已具备资质,以及物料是否被分配了适当的有效期或复验期。该审查还应确定来料控制措施是否足以防止使用不合格的组分、容器和密封件。
A description of how you will test each component lot for conformity with all appropriate specifications for identity, strength, quality, and purity. If you intend to accept any results from your supplier's COA instead of testing each component lot for strength, quality, and purity, specify how you will robustly establish the reliability of your supplier's results through initial validation as well as periodic revalidation. In addition, include a commitment to conduct at least one specific identity test for each incoming component lot as required.
说明你们将如何测试每个组分批次是否符合所有适当的鉴别、含量、质量和纯度规格。如果你们打算接受供应商的COA结果来代替对每个组分批次进行含量、质量和纯度测试,请具体说明你们将如何通过初次验证以及定期再验证,可靠地确定供应商结果的可靠性。此外,需包含一项承诺,即按要求对每个来料组分批次进行至少一项特定的鉴别测试。
Identity test results of raw materials currently in inventory. In the instance of no remaining raw material is available or within expiry, provide the test results for methanol that may be present in your finished OTC drug products.
当前库存原材料的鉴别检测结果。若无剩余原材料可用或已超出有效期,请提供可能存在于你们公司成品OTC药品中的甲醇测试结果。
2. Your firm failed to establish and follow adequate written procedures for cleaning and maintenance of equipment (21 CFR 211.67(b)).
你们公司未能建立并遵循充分的设备清洁和维护书面程序(21 CFR 211.67(b))。
You lacked the cleaning validation studies required by your written procedures for OTC drug products. You failed to demonstrate your cleaning practices are adequate to remove potential contaminants and to prevent product carry over in shared equipment (for example, fillers and small utensils used for formulation) used to manufacture your OTC drug products.
你们缺乏你们公司OTC药品书面程序所要求的清洁验证研究。你们公司未能证明你们公司的清洁操作足以去除潜在污染物,并防止在用于生产你们公司OTC产品的共用设备(例如,灌装机和用于配制的小型器具)中出现产品残留。
Inadequate removal of residues from manufacturing equipment during cleaning can lead to the contamination of drug products subsequently manufactured on nondedicated equipment.
清洁过程中未能充分去除生产设备上的残留物,可能导致后续在非专用设备上生产的药品受到污染。
In your response, you commit to conducting cleaning verifications for the removal of residues left by cleaning chemicals and organic materials. Specifically, you commit to completing a cleaning validation by the end of Q4 of 2025.
在你们的回复中,你们承诺将对清洁化学品和有机物质残留的清除情况进行清洁验证。具体而言,你们承诺在2025年第四季度末前完成清洁验证。
Your response is inadequate. You fail to provide specific details, such as predetermined acceptance criteria, cleaning frequency, and procedures and acceptable holding time limit for dirty equipment, supported by cleaning validation. Furthermore, you fail to assess the impact of your inadequate cleaning processes on product that is currently on the market and within expiry. Of note, cleaning deficiencies were discussed with your firm in a previous FDA inspection.
你们的回复不充分。你们未能提供由清洁验证支持的具体细节,例如预先确定的验收标准、清洁频率、经清洁验证支持设备的处理程序及可接受的最长存放时间限制。此外,你们未能评估清洁工艺不足对当前市场上且在有效期内的产品的影响。值得注意的是,清洁缺陷在之前的FDA检查中已与你们讨论过。
In response to this letter, provide:
作为对本信的回应,请提供:
An interim protocol for your cleaning verification with acceptance criteria, sampling methods, locations, and other considerations relevant to your equipment cleaning program.
一项关于你们清洁确认的临时规程,包含验收标准、取样方法、取样位置以及与设备清洁计划相关的其他注意事项。
A corrective action and preventive action (CAPA) plan, based on the retrospective assessment of your cleaning program, that includes appropriate remediations to your cleaning processes and practices and timelines for completion. Provide a detailed summary of vulnerabilities in your process for lifecycle management of equipment cleaning. Describe improvements to your cleaning program, including enhancements to cleaning effectiveness; improved ongoing verification of proper cleaning execution for all products and equipment; and all other needed remediations.
基于对你们清洁计划回顾性评估的纠正与预防措施(CAPA)计划,包括对你们清洁工艺和实践的适当整改措施及完成时间表。提供一份关于设备清洁生命周期管理流程中薄弱环节的详细总结。描述对清洁计划的改进,包括提高清洁效果;改进对所有产品和设备正确清洁执行的持续确认;以及所有其他必要的整改措施。
Appropriate improvements to your cleaning validation program, with special emphasis on incorporating conditions identified as worst case in your drug manufacturing operation. This should include but not be limited to, identification and evaluation of all worst-case:
对你们公司清洁验证计划的适当改进,特别强调纳入在你们公司药品生产运营中被确定为最差情况的条件。这应包括但不限于,对所有最差情况的识别和评估:
Drugs of lower solubility in their cleaning solvents
在清洁溶剂中溶解度较低的药品
Drugs with characteristics that make them difficult to clean
具有难以清洁特性的药品
Swabbing locations for areas that are most difficult to clean
最难以清洁区域的擦拭取样位置
Maximum hold times before cleaning
清洁前的最长保留时间
In addition, describe the steps that must be taken in your change management system before introduction of new manufacturing equipment or a new product.
此外,描述在引入新生产设备或新产品之前,必须在变更管理系统中采取的步骤。
A summary of updated SOPs that ensure an appropriate program is in place for verification and validation of cleaning procedures for products, processes, and equipment.
更新后的标准操作规程(SOP)摘要,以确保制定有适当的计划用于对产品、工艺和设备的清洁程序进行确认和验证。
A detailed risk assessment addressing the hazards posed by distributing drug products (such as (b)(4)) manufactured using shared equipment that may have been improperly cleaned. Specify actions you will take in response to the risk assessment, such as customer notifications and product recalls.
一份详细的风险评估,以解决使用可能清洁不当的共用设备生产的药品(例如 (b)(4))分销所带来的危害。具体说明为应对该风险评估将采取的行动,例如客户通知和产品召回。
3. Your firm failed to establish adequate written procedures for production and process control designed to assure that the drug products you manufacture have the identity, strength, quality, and purity they purport or are represented to possess (21 CFR 211.100(a)).
你们未能建立充分的生产和工艺控制书面程序,以确保你们生产的药品具有其声称或标明的特性、规格、质量和纯度(21 CFR 211.100(a))。
Your firm failed to conduct process validations for all your over-the-counter (OTC) drug products, including but not limited to your (b)(4) products. Our investigators also found three instances of bulk lot (b)(4) for your (b)(4) due to out of specification results.
你们未能对所有非处方(OTC)药品进行工艺验证,包括但不限于你们的(b)(4)产品。我们的调查员还发现,你们的(b)(4)有三个大货批次因(b)(4)导致检测结果超标。
Process validation evaluates the soundness of design and the state of control of a process throughout its lifecycle. Each significant stage of a manufacturing process must be designed appropriately and assure the quality of raw material inputs, in-process materials, and finished drugs. Process qualification studies determine whether an initial state of control has been established. Successful process qualification studies are necessary before commercial distribution. Thereafter, ongoing vigilant oversight of process performance and product quality is necessary to ensure that you maintain a stable manufacturing operation throughout the product lifecycle. See FDA's guidance for industry Process Validation: General Principles and Practices at for general principles and approaches that the FDA considers appropriate elements of process validation.
工艺验证评估工艺在其整个生命周期中设计的合理性和受控状态。生产工艺的每个重要阶段都必须进行适当设计,并确保原材料输入、中间体和成品药品的质量。工艺确认研究确定是否已建立初始的受控状态。成功的工艺确认研究是商业分销前所必需的。此后,需要对工艺性能和产品质量进行持续、严格的监督,以确保在整个产品生命周期中保持稳定的生产操作。关于FDA认为属于工艺验证适当要素的一般原则和方法,请参阅FDA行业指南《工艺验证:一般原则与实践》,网址为
In your response, you state that you will perform an evaluation and create a gap analysis to prioritize the completion of products and formulations requiring validation. You commit to have a validation protocol completed before the next scheduled run on nonvalidated products, so the validations can be conducted concurrently.
在你们的回复中,你们表示将进行评估并创建差距分析,以优先完成需要验证的产品和配方的验证工作。你们承诺在下一个非验证产品的计划生产运行之前完成验证方案,以便可以同时进行验证。
Your response is inadequate. You did not provide an impact assessment on the drugs you distributed without completing validation studies to ensure acceptable quality of your drug products.
你们的回复不够充分。你们未提供对未完成验证研究以确保药品质量可接受即已分销药品的影响评估。
Additionally, based on inspection findings, your (b)(4) system was not in a state of control because of inadequate testing and monitoring and inadequate sanitization. You use (b)(4) as a component in your drug products and for equipment cleaning. (b)(4) for pharmaceutical purposes must be suitable for its intended use and must be routinely tested to ensure ongoing conformance with appropriate chemical and microbiological attributes. The lack of adequate monitoring provides no assurance the system performs as intended.
此外,根据检查发现,你们的(b)(4)系统由于测试和监控不足以及清洁不充分,未处于受控状态。你们将(b)(4)用作药品的组分并用于设备清洁。用于制药目的的(b)(4)必须适合其预期用途,并且必须进行常规测试,以确保其持续符合适当的化学和微生物属性。缺乏充分的监测无法保证该系统按预期运行。
In your response, you commit to adding additional testing points and reinstating the (b)(4) testing frequency. You also commit to monitoring and documenting the water system maintenance performed by the contractor.
在你们的回复中,你们承诺增加额外的测试点并恢复(b)(4)的检测频率。你们还承诺监控并记录承包商执行的供水系统维护工作。
Your response is inadequate. You fail to address the effects on the product resulting from the use of (b)(4) that is not adequately tested and monitored for its intended use.
你们的回复不够充分。你们未能解决使用未经充分测试和监测以确保其预期用途的(b)(4)对产品造成的影响。
In response to this letter, provide:
作为对本信的回应,请提供:
A detailed summary of your validation program for ensuring a state of control throughout the product lifecycle, along with associated procedures. Describe your program for process performance qualification and ongoing monitoring of both intrabatch and interbatch variation to ensure a continuing state of control.
一份关于你们验证计划的详细总结,以确保在整个产品生命周期中保持受控状态,并附上相关程序。描述你们用于工艺性能确认以及持续监控批内和批间变异以确保持续受控状态的计划。
A timeline for performing process performance qualification for each of your marketed drug products.
对你们每种已上市药品进行工艺性能确认的时间表。
Process performance protocol(s) and written procedures for qualification of equipment and facilities.
用于设备和设施确认的工艺性能方案和书面程序。
A procedure for your (b)(4) system monitoring that specifies routine microbial testing of (b)(4) to ensure its acceptability for use in each batch of drug products produced by your firm.
你们(b)(4)系统监控程序,规定对(b)(4)进行常规微生物检测,以确保其适用于你们公司生产的每批药品。
The current action/alert limits for total counts and objectionable organisms used for your (b)(4) system.
你们(b)(4)系统当前使用的总菌数和特定微生物的行动限/警戒限。
A procedure governing your program for ongoing control, maintenance, and monitoring that ensures the remediated system consistently produces (b)(4) that meets (b)(4) monograph specifications and appropriate microbial limits.
一个管理你们公司的持续控制、维护和监控计划的程序,以确保经整改后的系统能持续生产出符合(b)(4)各论规格及适当微生物限度的(b)(4)。
4. Your firm failed to establish adequate written responsibilities and procedures applicable to the quality control unit and to follow such written procedures (21 CFR 211.22 (d)).
你们未能建立适用于质量控制部门的充分书面职责和程序,且未能遵循此类书面程序(21 CFR 211.22 (d))。
You lacked adequate quality unit (QU) oversight for the manufacture of your OTC drug products. For example:
你们缺乏对OTC药品生产的充分质量部门(QU)监督。例如:
You have not performed an annual product review (APR) of your OTC drug products manufactured since 2022.
你们未对自2022年以来生产的OTC药品进行年度产品回顾(APR)。
You failed to follow your procedure for raw material sampling, in that multiple different materials were held in the sampling area, which poses the risk of potential contamination and mix-ups.
你们未能遵循你们的原材料取样程序,因为取样区存放了多种不同的物料,这带来了潜在污染和混淆的风险。
An adequate QU overseeing all manufacturing operations is necessary to consistently ensure drug quality. Your firm's quality systems are inadequate. See FDA's guidance document Quality Systems Approach to Pharmaceutical CGMP Regulations at for help in implementing quality systems and risk management approaches to meet the requirements of CGMP regulations (21 CFR parts 210 and 211).
一个充分的质量部门(QU)监督所有生产操作对于持续确保药品质量是必要的。你们公司的质量体系不足。关于实施质量体系和风险管理方法以满足CGMP法规(21 CFR 210和211部分)要求的帮助,请参阅FDA指南文件《药品CGMP法规的质量体系方法》,网址为
In your response, you commit to ensuring that procedures are followed and that programs are up to date and complete going forward.
在你们的回复中,你们承诺确保遵循程序,并确保未来的计划得到更新和完善。
Your response is inadequate. Although you commit to addressing the observations we identified, your response lacks sufficient details about the systemic remediations needed for your QU and your quality system.
你们的回复不够充分。尽管你们承诺解决我们发现的问题,但你们的回复缺乏关于你们公司QU和质量体系所需系统性整改措施的足够细节。
In response to this letter, provide:
作为对本信的回应,请提供:
A comprehensive assessment and remediation plan to ensure your QU is given the authority and resources to effectively function. The assessment should also include but not be limited to:
一项全面的评估和整改计划,以确保你们公司的QU被赋予有效履行职责的权力和资源。该评估还应包括但不限于:
A determination of whether procedures used by your firm are robust and appropriate.
确定你们公司使用的程序是否健全且适当。
Provisions for QU oversight throughout your operations, to evaluate adherence to appropriate practices.
规定QU在整个运营过程中的监督职责,以评估对适当规范的遵守情况。
A complete and final review of each batch and its related information before the QU disposition decision.
在QU做出放行决定前,对每个批次及其相关信息进行完整和最终的审核。
Oversight and approval of investigations and discharging of all other QU duties to ensure identity, strength, quality, and purity of all products.
对调查的监督和批准,以及履行所有其他QU职责,以确保所有产品的鉴别、含量、质量和纯度。
CGMP Consultant Recommended
建议聘请CGMP顾问
Based upon the nature of the violations we identified at your firm, and because you failed to correct repeat violations, we strongly recommend that your firm engage a consultant qualified as set forth in 21 CFR 211.34 to assist your firm in meeting CGMP requirements.
基于我们在你们公司发现的违规行为的性质,以及你们未能纠正重复违规行为,我们强烈建议你们聘请符合21 CFR 211.34规定的合格顾问,以协助你们公司满足CGMP要求。
Your use of a consultant does not relieve your firm's obligation to comply with CGMP. Your firm's executive management remains responsible for resolving all deficiencies and systemic flaws to ensure ongoing CGMP compliance.
你们使用顾问并不能免除你们公司遵守CGMP的义务。你们的高级管理层仍有责任解决所有缺陷和系统性缺陷,以确保持续符合CGMP。
Conclusion
结论
The violations cited in this letter are not intended to be an all-inclusive list of violations that exist at your facility. You are responsible for investigating and determining the causes of any violations and for preventing their recurrence or the occurrence of other violations.
本信中引用的违规行为并非意在列出你们公司设施存在的所有违规行为。你们公司有责任调查和确定任何违规行为的原因,并防止其再次发生或其他违规行为的发生。
Correct any violations promptly. Failure to address this matter promptly and adequately may result in regulatory or legal action without further notice, including, without limitation, seizure and injunction. Unresolved violations may also prevent other Federal agencies from awarding contracts.
请及时纠正任何违规行为。若未能及时、充分地解决此事,可能导致监管或法律行动,恕不另行通知,包括但不限于查封和禁令。未解决的违规行为也可能妨碍其他联邦机构授予合同。
Failure to address violations may also cause FDA to withhold issuance of Export Certificates. FDA may withhold approval of new applications or supplements listing your firm as a drug manufacturer until any violations are completely addressed and we confirm your compliance with CGMP. We may re-inspect to verify that you have completed corrective actions to address any violations.
未能解决违规行为也可能导致FDA暂停签发出口证书。FDA可能会暂扣将你们公司列为药品生产商的新申请或补充申请的批准,直到所有违规行为完全解决且我们确认你们公司符合CGMP为止。我们可能会重新检查以核实你们公司已完成解决任何违规行为的纠正措施。
This letter notifies you of our findings and provides you an opportunity to address the above deficiencies. After you receive this letter, respond to this office in writing within 15 working days. Specify what you have done to address any violations and to prevent their recurrence. In response to this letter, you may provide additional information for our consideration as we continue to assess your activities and practices. If you cannot complete corrective actions within 15 working days, state your reasons for delay and your schedule for completion.
本信通知你们公司我们的发现,并为你们公司提供解决上述缺陷的机会。你们公司收到此信后,请在15个工作日内以书面形式回复本办公室。具体说明你们公司为解决任何违规行为并防止其再次发生所采取的措施。作为对本信的回应,你们公司可以提供更多信息供我们考虑,同时我们将继续评估你们公司的活动和实践。如果你们公司无法在15个工作日内完成纠正措施,请说明延迟原因和完成时间表。
Send your electronic reply to CDER-OC-OMQ-Communications@fda.hhs.gov. Identify your response with FEI 3007953761 and ATTN: Joan Johnson.
请将您的电子回复发送至CDER-OC-OMQ-Communications@fda.hhs.gov。请在回复中注明FEI 3007953761并注明由Joan Johnson收阅。
-END-
2026药典符合性实验室管理和分析方法验证及数据统计分析专题研讨班
时间:2026年03月26日-28日
地点:上海市
课程背景与目标
课程时间安排
03月27日 9:00-12:00 13:30-16:30
符合2025版药典的实验室规范化管理
一、实验室质量管理法规体系概述
二、符合2025版药典的药品检验流程管理
1.人员培训和资质确认(职责及培训计划、上岗前培训与资质确认、培训档案管理)
2.2025版药典:药品取样全流程合规要点(风险控制点:环境、工具、交叉污染)
3.样品管理(法规、转运和接受、储存与发放)
4.2025版药典《9094分析仪器确证指导原则》:基于风险评价分析仪器分类管理与生命周期管理(分析仪器分类、编号管理、校验与维护)
5.通则《0291国家药品标准物质通则》、通则《9901国家药品标准物质研制指导原则》增修订内容及实施要点
a通则0291 国家药品标准物质通则修订及实施转化解读
b通则9901国家药品标准物质研制指导原则修订及实施转化解读
药典凡例与药典符合性检验技术操作规范
一、2025版药典凡例深度解读与合规应用
1.凡例的法律地位与核心作用(作为药典标准解读的“纲领性文件”,指导检验全流程)
2.药典凡例法定约束力边界
3.术语定义(如 “精密称定”“恒重”“澄清” 等术语的量化标准与操作边界)
4.标准表述规则(如“不得过”“应符合规定” 等表述的合规判定逻辑)
二、药典核心检验方法原理与与符合性操作
1.常用物理常数测定方法(熔点、沸点、比旋度)的药典要求与操作规范
2.鉴别试验方法实操要点标准操作
3.杂质检查方法核心原理,如杂质限量计算、外标法/内标法应用场景,有关物质检测的色谱
4.含量测定方法分类解析,容量分析法、紫外分光光度法、色谱法的适用范围与选择逻辑。
主讲人:张老师
03月28日 9:00-12:00 13:30-16:30
一、基于ICHQ2(R2)、中国药典9101与USP1225的分析方法验证综合考量
1)方法验证基本要求
1 分析方法验证适用范围
2.分析方法的生命周期管理
3 可报告范围确定
4 稳定性指示方法
5 多变量分析方法
2)分析方法验证的主要内容及关键要点
1专属性评估,多种方法验证方法的专属性
2准确度实验设计原则和可接受标准
3精密度实验设计原则和可接受标准
4 工作范围确定
4.1 校准模型建立 线性或非线性和范围设计的基本原则
4.2 低范围限的验证,检测限和定量限的评估
5方法耐用性的设计和评价
6系统适用性试验的选择
分析方法评价和使用中的统计学工具
一、测量不确定度及其应用
1.测量不确定度的应用
●计量认证、计量确认、质量认证和实验室认可
●参加能力验证、或国际间实验比对活动
●标准物质标定
●仪器校准和检定
●判定检测结果符合性,特别对临界值的判断时
●其它情况,如客户有要求时
2.检测结果符合性评价
3.置信区间的确定-案例剖析
二、异常值的检验与剔除(USP<1010>)
1.什么样的数据必须剔除?
2.几种常用判别分析异常数据的统计方法?
●拉依塔准则
●格罗布斯准则
●狄克逊准则
三、分析结果的比较
1.分析结果比对要点
●人员比对●仪器比对●方法比对
●标准物质比对●留样再试●实验室间比对
2.统计分析比对试验结果方法
●F检验法(USP推荐方法)
●T检验法(USP推荐方法)
●En值判断法
●CD值判断法
●允差判断法
●Z比分数
●F检验法的案例分析
四、Horwitz方程及其应用
1.实验室间数据偏差评价
2.Horwitz方程在比对试验质量评估中的应用
五、如何评价分析方法的准确度和精密度(USP<1210>)
(1)基于测量不确定度的考虑
(2)常量分析方法回收率置信区间
(3)精密度的置信区间
(4)微量分析方法置信区间
(5)准确度与精密度的联合验证
六、检测限和定量限的评估新方法(USP<1210>)
主讲人:王老师
会务费
培训费:4800元/单位,每单位限额3人(1600元/人),包含(专家费、资料费、场地费、现场问答等)。食宿统一安排,费用自理。
培训报名联系人:王老师
微信/手机:155 0614 0531

扫码添加微信
【点这里,进入医药数据库,搜索下载原文】

药学检测服务推荐
一、化药检测服务
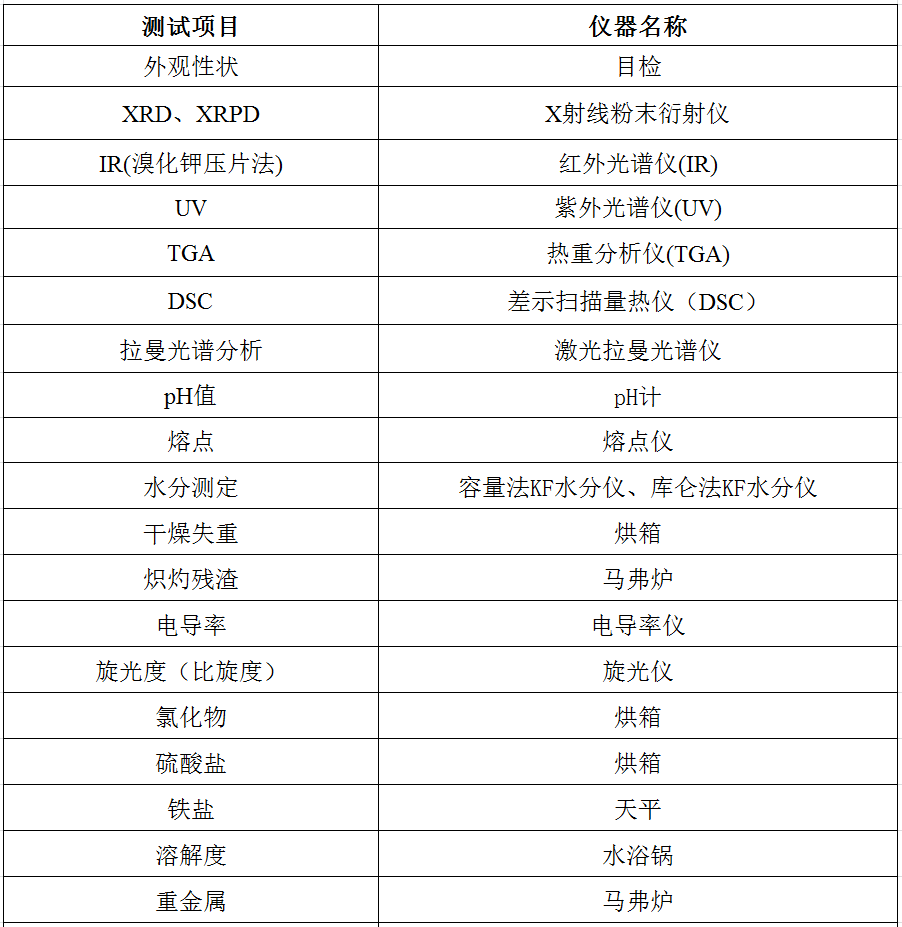



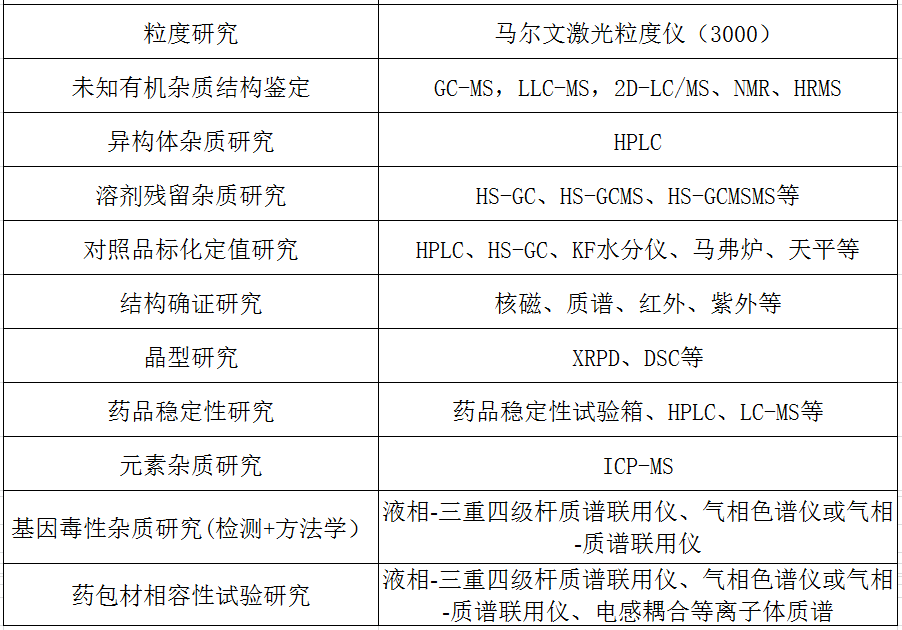
二、化药质量研究服务

三、生物药检测服务

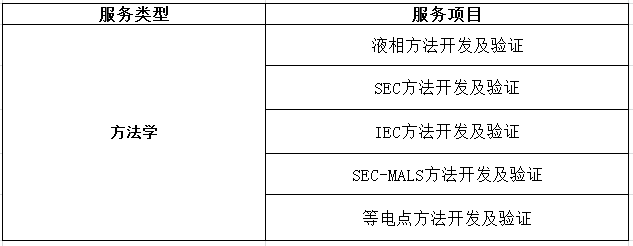
四、生物药质量研究服务
五、药品毒理安全性评价服务

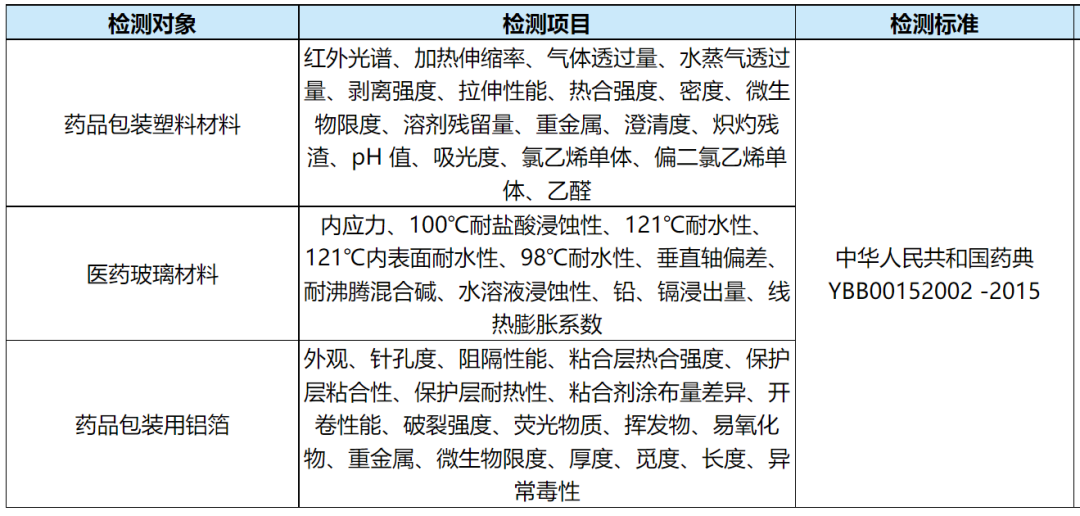
六、药品包装材料理化检测
实验室资质:CMA、CNAS
实验室所在地:上海、苏州、青岛、广州、福州
联系我们:151-9002-7486/181-0068-0718(微信同号)


文章来源:科威利华,本公众号所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,不希望被转载的单位、媒体或个人可与我们联系(274190388@qq.com),我们将立即进行删除处理。所有文章仅代表作者观点,不代表本站立场。






