 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库【JMC】中国药科大学谢唯佳/吴亮团队:新型α-葡萄糖苷酶抑制剂的设计及抗糖尿病活性研究

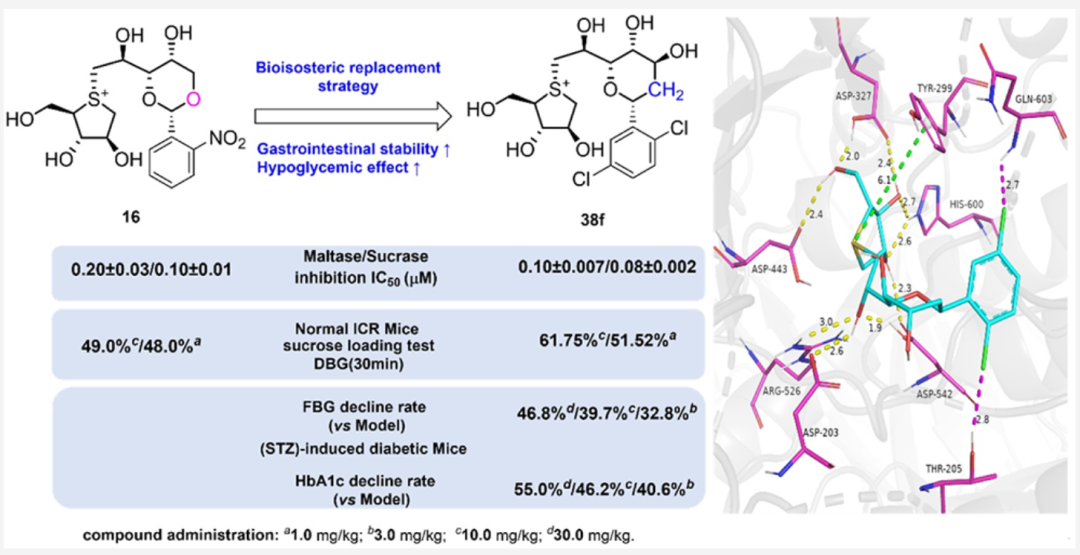
近日,中国药科大学谢唯佳/吴亮团队在药物化学权威期刊Journal of Medicinal Chemistry上发表题为"靶向α-葡萄糖苷酶的新型C-糖基吡喃型锍盐与硒鎓盐的发现及其强效抗高血糖活性研究"的研究论文,报道了一类酸稳定的C-糖基吡喃型锍盐类α-葡萄糖苷酶抑制剂。针对天然锍盐先导化合物中苄叉缩醛基团胃酸不稳定的问题,研究团队基于生物电子等排体原理,将其改造为C-糖基吡喃骨架,设计合成了系列锍盐与硒鎓盐衍生物。其中,化合物38f(2,5-二氯苯基取代锍盐)对大鼠肠道麦芽酶和蔗糖酶的IC₅₀分别为0.10 μM和0.08 μM,活性显著优于临床药物阿卡波糖和伏格列波糖。38f在模拟胃液中2小时降解率仅1.1%,模拟肠液中4小时保持99%以上完整,展现出卓越的胃肠道稳定性。STZ糖尿病小鼠模型中,3 mg/kg剂量即可降低空腹血糖32.8%,优于阿卡波糖50 mg/kg的24.3%降幅,且能显著改善肝糖原和HbA1c水平。14天毒性研究证实其安全性良好。分子对接揭示38f通过阳离子-π作用与Tyr299结合,双氯原子形成卤键双锚定点,实现高亲和力抑制。该研究为开发新一代酸稳定、高效低毒的α-葡萄糖苷酶抑制剂提供了重要先导化合物。
1 min速览
研究背景
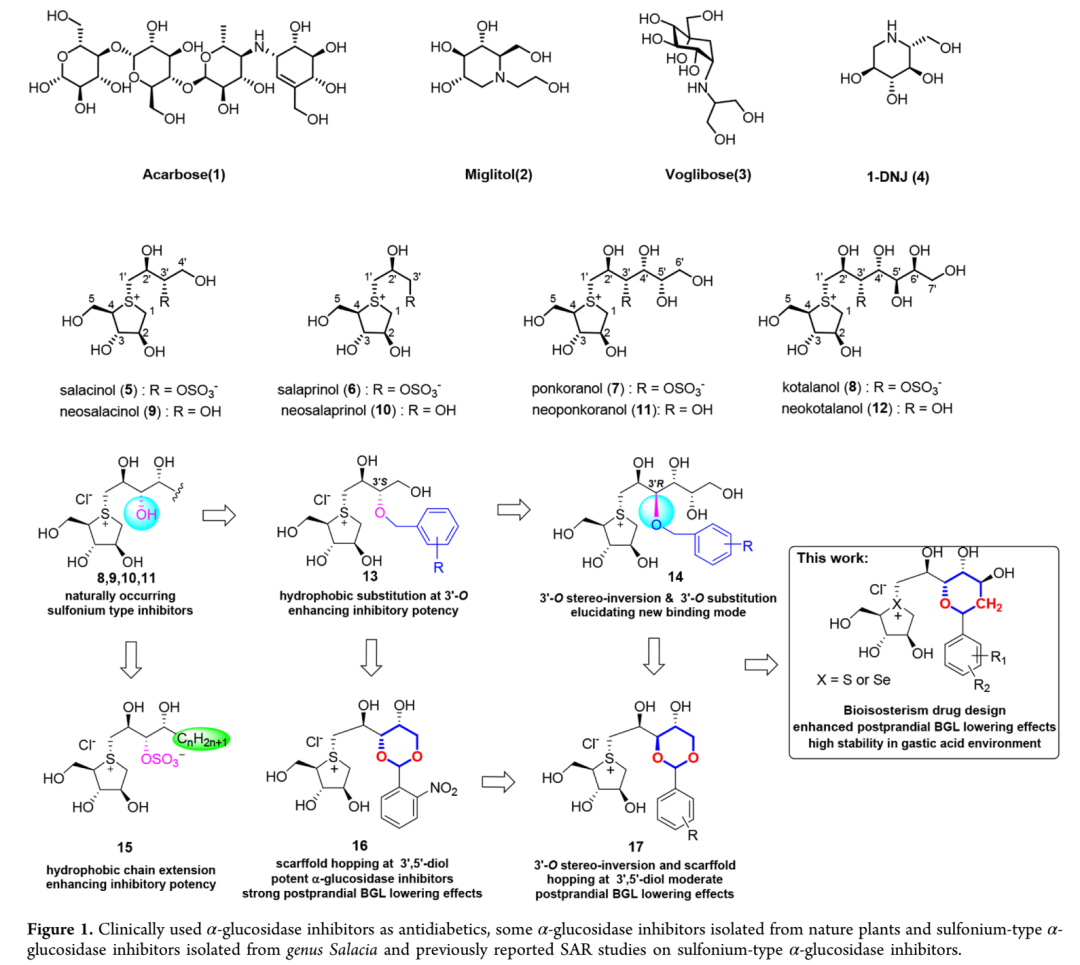
2型糖尿病(T2DM)已成为全球第三大死因,α-葡萄糖苷酶抑制剂通过延缓碳水化合物水解有效控制餐后高血糖,是亚洲高碳水饮食人群的重要治疗选择。临床常用药物阿卡波糖、伏格列波糖存在胃肠道副作用大、疗效有限等缺陷。天然来源的锍盐类化合物(如Salacia属植物中的neoponkoranol)虽活性优异且安全性高,但其苄叉缩醛结构在胃酸环境中不稳定,导致药效损失。因此,开发兼具强效抑制活性与胃酸稳定性的新型α-葡萄糖苷酶抑制剂具有重要临床价值。
重点内容
1. 分子设计策略:基于生物电子等排体原理,将酸敏感的苄叉缩醛改造为水解稳定的C-糖基吡喃骨架,保留关键药效团(五元锍环、2S,3S,4R立体构型、多羟基侧链),并引入系列吸电子取代基(卤素、三氟甲基等)优化活性。
2. 关键化合物发现:化合物38f(2,5-二氯苯基取代锍盐)活性最优:对大鼠麦芽酶和蔗糖酶的IC₅₀分别为0.10 μM和0.08 μM,较阿卡波糖提升30倍以上。Hammett分析证实吸电子取代基显著增强麦芽酶抑制活性(ρ = +1.61)。
3. 稳定性突破:38f在模拟胃液(pH 1.3)中2小时降解率仅1.1%(对照化合物41降解68.8%),肠液中4小时保持>99%完整,彻底解决酸不稳定难题。
4. 体内药效验证:STZ糖尿病小鼠中,38f以3 mg/kg剂量降低空腹血糖32.8%,优于阿卡波糖50 mg/kg的24.3%;OGTT、ITT及糖耐量试验均显示剂量依赖性优异降糖效果,并显著改善肝糖原与HbA1c。
5. 安全性评价:14天亚急性毒性研究(≤500 mg/kg)未见肝肾毒性,治疗窗宽广。
6. 作用机制阐明:动力学研究证实38f为竞争性抑制剂(Ki: 0.31/0.27 μM);分子对接揭示锍阳离子与Tyr299的阳离子-π作用及双氯原子的卤键双锚定为关键结合模式。
研究总结
本研究通过C-糖基吡喃骨架替换策略,成功解决了天然锍盐类α-葡萄糖苷酶抑制剂的酸稳定性缺陷,获得兼具强效活性、胃酸稳定、优异体内药效及良好安全性的候选化合物38f。该工作为开发新一代抗糖尿病药物提供了重要先导化合物,也为糖模拟物类药物的理性设计提供了新思路。
图片来源:ACS
详细阅读
01
INTRODUCTION
研究背景
糖尿病已成为全球第三大死因,2型糖尿病占全部病例的90%以上。α-葡萄糖苷酶抑制剂通过延缓肠道碳水化合物水解降低餐后血糖,对高碳水饮食人群尤为适用。目前临床使用的阿卡波糖、米格列醇和伏格列波糖存在胃肠道不良反应和疗效有限等问题。天然来源的锍盐类化合物(如从Salacia属植物中分离的化合物5-12)具有优异的α-葡萄糖苷酶抑制活性和高安全性,其中1-脱氧野尻霉素已成为首个获批的植物源α-葡萄糖苷酶抑制剂。前期研究表明,五元锍环的2S,3S,4R立体构型、侧链的2'S-OH和4'R-OH协同配置对活性至关重要,而苄叉缩醛保护基团的酸不稳定性限制了其在胃部的稳定性——作者推测在小肠发挥作用的活性物种可能包括原型药物及其酸催化降解产物,后者活性显著降低。因此,本研究旨在通过将苄叉缩醛转化为C-糖基吡喃骨架,设计酸稳定的下一代α-葡萄糖苷酶抑制剂,以确保原型药物完整到达肠道作用部位,实现最大治疗指数。
02
Drug Design
①设计策略与化学合成
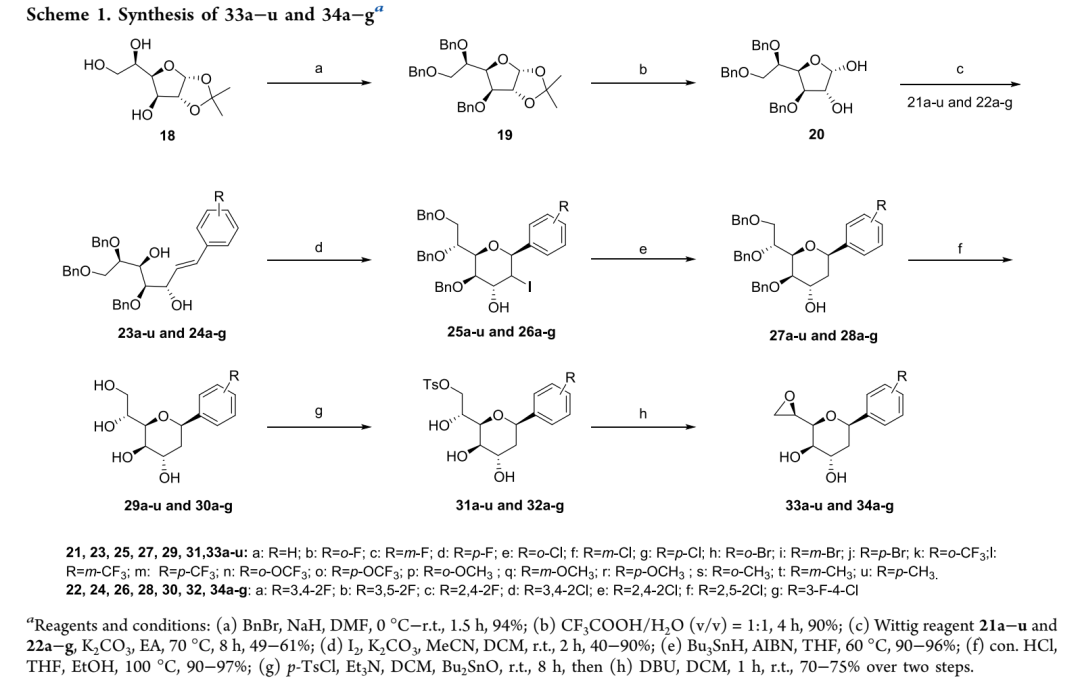
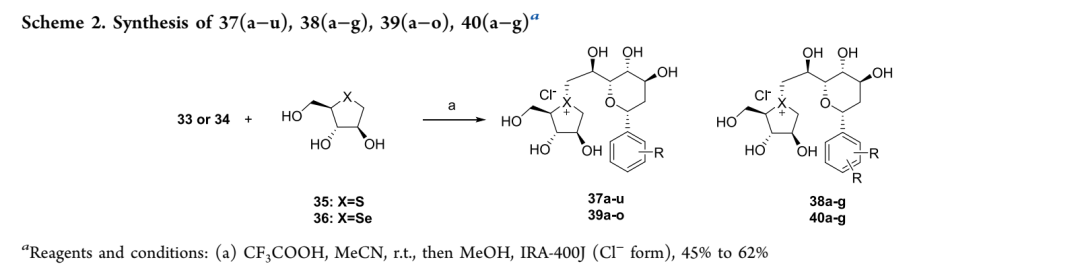
本研究旨在开发兼具强效生物活性与胃酸稳定性的新一代α-葡萄糖苷酶抑制剂。为从根本上提升目标分子在上消化道的化学稳定性,同时保持侧链必要的构象与亲水性,研究者保留了前期报道化合物16的主骨架,将酸不稳定的苄叉缩醛中的氧原子替换为亚甲基碳,转化为水解稳定的C-糖基吡喃骨架。初步对接研究表明,该设计与靶标N端麦芽酶-葡萄糖淀粉酶(ntMGAM)的结合模式与前期锍盐类化合物相似。基于前期构效关系研究,进一步拓展结构多样性:一是考察苄叉苯环上不同取代基对活性与选择性的影响;二是受硒糖类化合物作为新兴血糖调节糖模拟物的报道启发,依据生物电子等排体原理将锍阳离子中心替换为硒鎓基团,构建系列硒鎓盐衍生物。关键环氧化物中间体33a-u和34a-g的合成以1,2-O-异亚丙基-α-D-葡萄糖呋喃糖为起始原料,经苄基保护、异亚丙基脱除、Wittig反应、碘介导环化、自由基脱碘、脱苄基、对甲苯磺酰化及DBU促进的环氧化等步骤制得;随后与硫糖35在催化量三氟乙酸作用下偶联,经阴离子交换树脂处理得到锍盐氯化物37和38,硒鎓盐39和40则通过类似方法由相应环氧化物合成。
②α-葡萄糖苷酶抑制活性评价
合成的锍盐和硒鎓盐类化合物经系统体外酶活评价,以天然锍盐neoponkoranol及临床药物伏格列波糖、阿卡波糖为对照,测定其对大鼠肠道α-葡萄糖苷酶(蔗糖酶和麦芽酶)的抑制活性。含苯基取代的化合物37a对麦芽酶和蔗糖酶的IC₅₀分别为0.95 μM和0.59 μM,表明侧链由苄叉缩醛转化为C-糖基吡喃结构未降低抑制活性。单取代衍生物中,卤素取代活性略有波动:氟或氯取代活性稍降或无显著变化(37b-37g),溴取代则中等增强活性(37h-37j);取代基位置对单取代衍生物活性影响较小,与前期报道不同。邻位三氟甲基取代的37k活性显著增强(麦芽酶IC₅₀ = 0.21 μM,蔗糖酶IC₅₀ = 0.10 μM),而间位和对位类似物活性稍降;邻位三氟甲氧基取代的37n亦为强效抑制剂,对位类似物37o活性略降。供电子基团(甲基、甲氧基)导致活性显著下降(37p-37u),表明缺电子苯环对强效抑制至关重要。双取代衍生物中,2,5-二氯苯基取代的38f活性最优(麦芽酶IC₅₀ = 0.10 μM,蔗糖酶IC₅₀ = 0.08 μM)。硒鎓盐类中,单取代苯基衍生物普遍较相应锍盐活性增强,如39d活性优于37d;但双取代硒鎓盐(40a-40g)未较相应锍盐进一步提升,40f活性甚至略低于38f。Hammett分析显示,单取代衍生物的麦芽酶抑制与σ常数呈显著正相关(R² = 0.52,ρ = +1.55,p < 0.01),扩展至单双取代衍生物后相关性增强(R² = 0.62,ρ = +1.61,p < 0.001),而蔗糖酶抑制对电子效应敏感性较低,提示两种酶抑制机制存在差异。

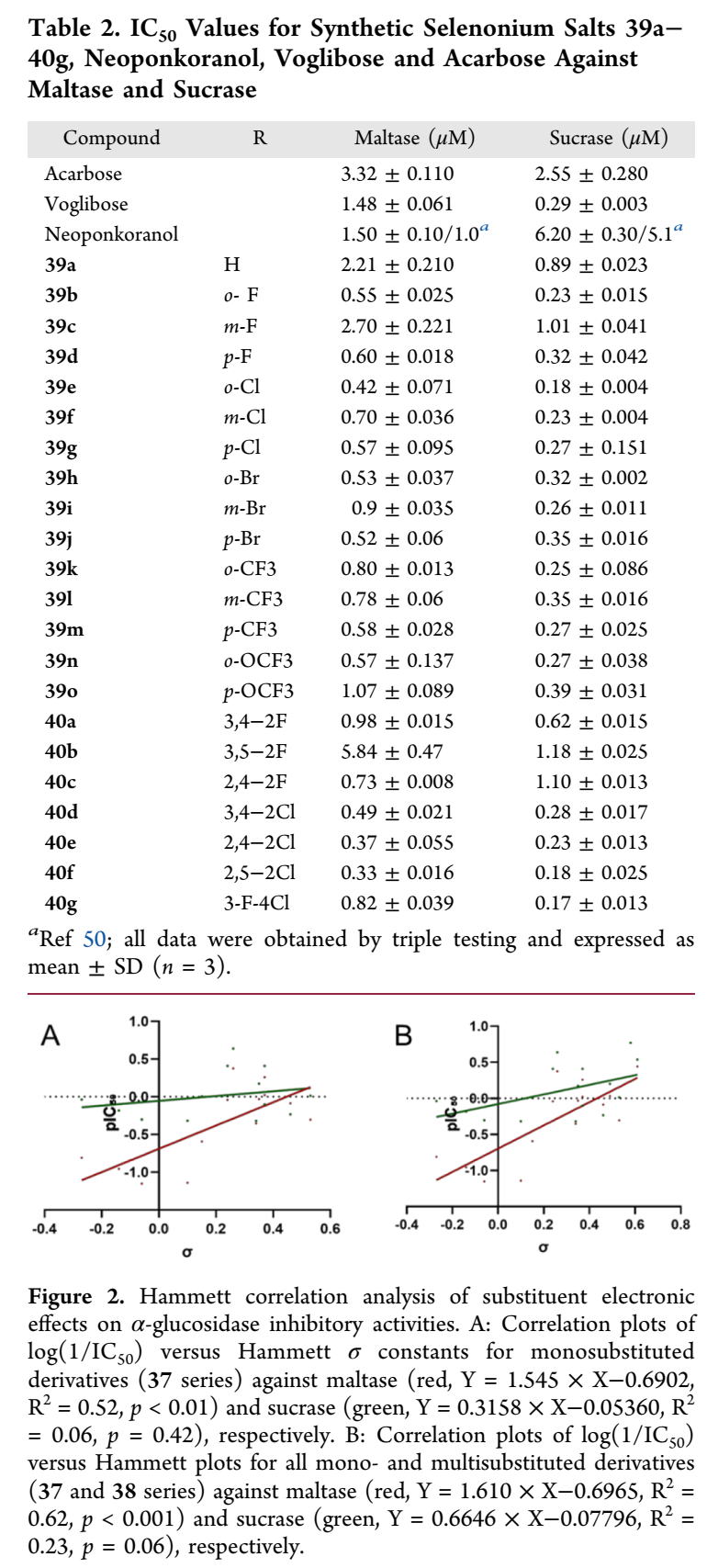
03
RESULTS
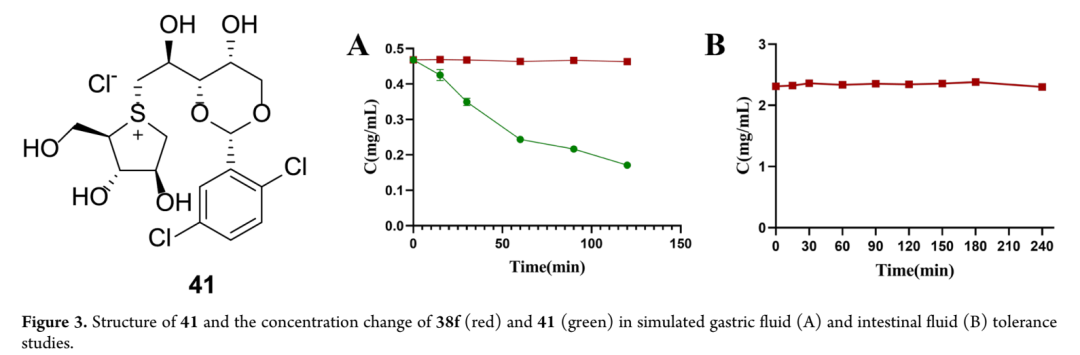
①胃液与肠液耐受性研究
为评价目标锍盐和硒鎓盐的胃酸稳定性,采用高效液相色谱法测定化合物38f与对照化合物41在人工胃液(USP,无菌,pH 1.3)中的稳定性。结果显示,38f在胃液中高度稳定,2小时孵育后降解率仅1.1%;而41在相同条件下呈时间依赖性显著降解:15分钟降解2%,30分钟25.4%,60分钟49.6%,120分钟达68.8%。该结果证实,以C-糖基吡喃替换苄叉缩醛成功解决了16的胃肠道酸不稳定性问题,使38f口服后能以完整形式大量到达肠道作用部位。进一步评价38f在模拟肠液(含胰酶和磷酸盐,无菌,pH 6.8)中的稳定性,发现其表现与胃液中相当,4小时孵育后原型药物剩余超过99%,超过典型小肠转运时间。38f在模拟胃液和肠液中的双重优异稳定性,为其口服药效提供了坚实的理化基础,确保治疗相关剂量的完整抑制剂到达小肠刷状缘膜发挥强效降糖作用。
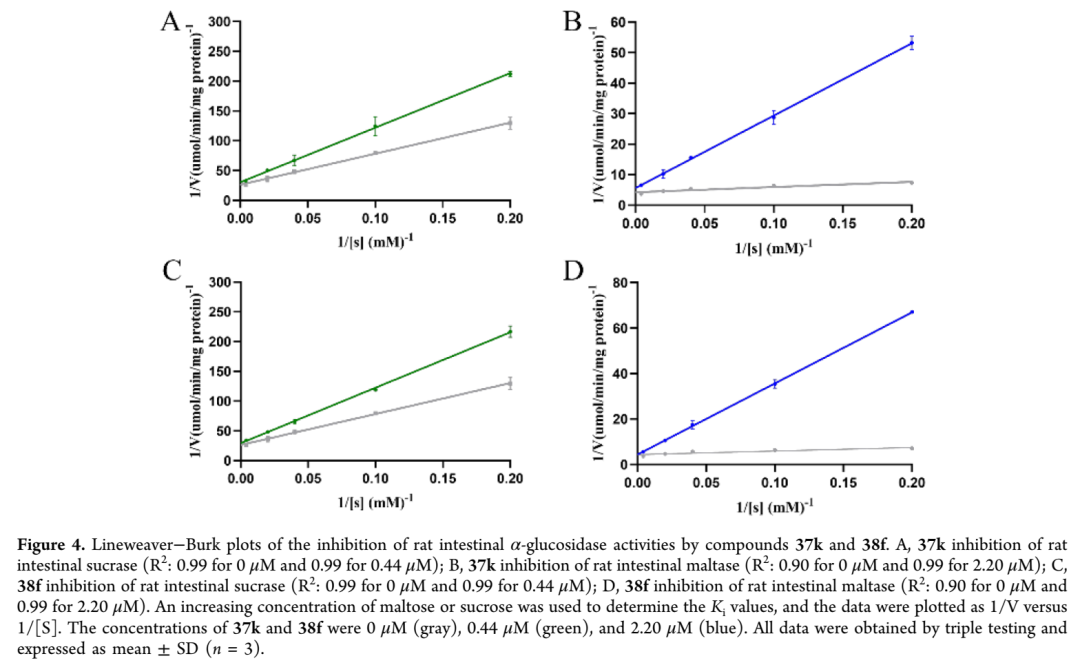
②酶动力学研究
为阐明该系列α-葡萄糖苷酶抑制剂的作用机制,对化合物37k和38f进行了Lineweaver-Burk酶动力学分析。采用小肠刷状缘膜囊泡来源的α-葡萄糖苷酶,以5-250 mM浓度的麦芽糖和蔗糖为底物进行测定。双倒数作图(1/V对1/[S])显示相交的线性回归,斜率随抑制剂浓度变化,表明二者通过活性位点结合产生竞争性抑制,高线性度支持竞争性抑制模型的可靠性。38f对麦芽酶和蔗糖酶的Ki值分别为0.31 μM和0.27 μM,结合亲和力优于37k(Ki: 0.79/0.31 μM)。通过Ki(麦芽酶)/Ki(蔗糖酶)比值计算选择性指数,38f的比值为0.31/0.27=1.14,提示其对蔗糖酶具有优先抑制倾向,该定量分析确认38f与临床对照药物具有不同的抑制特征,且与文献报道的天然α-葡萄糖苷酶抑制剂选择性指数大于1的趋势一致。

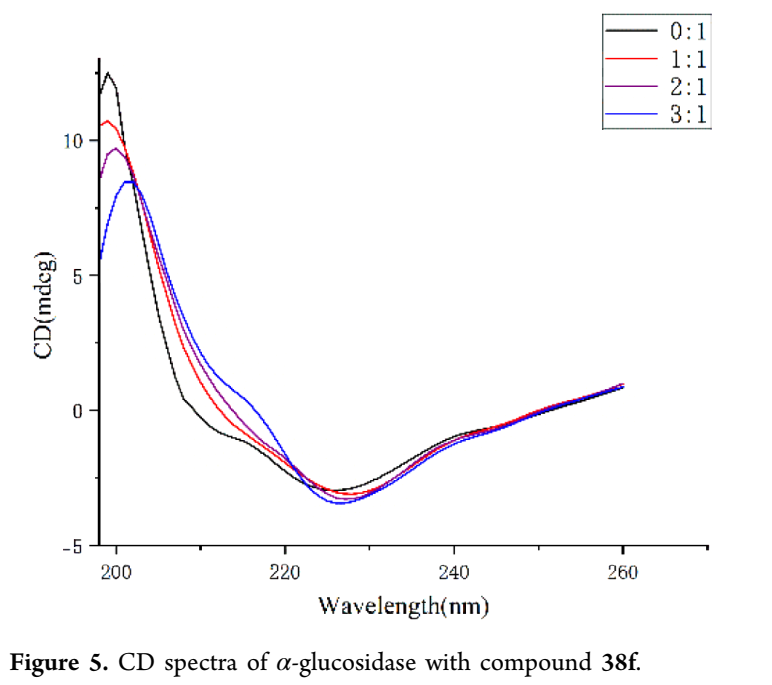
③圆二色谱分析
采用圆二色谱(CD)评价化合物38f对大鼠肠道α-葡萄糖苷酶复合物二级结构的影响。结果显示,天然α-葡萄糖苷酶在205-226 nm范围呈现特征信号,主要归因于其α-螺旋结构;经38f处理后,特征谱带的强度和形状发生明显改变,提示化合物诱导了酶的构象变化。二级结构的定量分析进一步证实,38f可引起肠道α-葡萄糖苷酶的构象重排,这可能与其催化活性下降相关。
④正常ICR小鼠的降血糖效应
为评价活性锍盐类α-葡萄糖苷酶抑制剂37k和38f在正常雄性ICR小鼠中的降血糖效果,进行了蔗糖负荷试验和麦芽糖负荷试验,并与neoponkoranol及临床药物阿卡波糖、伏格列波糖比较,同时设置前期化合物16作为对照。蔗糖负荷试验中,禁食12小时后灌胃蔗糖(2.5 g/kg)可快速升高血糖,30分钟达峰,120分钟恢复基线;候选化合物以10.0 mg/kg剂量给药后,37k和38f均强效抑制餐后血糖,早期效应优于阿卡波糖(20.0 mg/kg)和neoponkoranol(10.0 mg/kg),15分钟血糖降幅分别为51.7%和57.1%,30分钟为55.8%和61.8%,曲线下面积(AUC)分别降低47.1%和51.0%,而阿卡波糖仅降低40.5%。进一步以1.0 mg/kg低剂量评价,38f仍显示稳健降糖效果,15、30、60分钟血糖降幅分别为49.2%、51.5%、46.0%,AUC降低43.2%,优于阿卡波糖20.0 mg/kg的40.5%;37k在相同剂量下血糖控制效果亦与阿卡波糖相当。麦芽糖负荷试验中,禁食后灌胃麦芽糖(2.0 g/kg)使血糖30分钟达峰,1.0 mg/kg剂量的37k和38f均强效抑制餐后血糖,早期效应优于伏格列波糖(1.0 mg/kg)和neoponkoranol(1.0 mg/kg),30分钟血糖降幅分别为59.0%和57.8%,60分钟38f仍维持51.6%降幅,AUC分别降低46.9%和48.7%,优于伏格列波糖的39.8%和neoponkoranol的33.9%。与前期化合物16(体外活性与37k、38f相当)比较,在蔗糖负荷试验中16(1.0 mg/kg)表现略逊于阿卡波糖(20.0 mg/kg),而新设计化合物37k和38f(1.0 mg/kg)体内降糖效果显著增强,38f在所有时间点及AUC指标上均一致优于阿卡波糖和16;麦芽糖负荷试验中观察到相似趋势,38f展现出独特且显著的优势。对于局部起效的强效α-葡萄糖苷酶抑制剂,口服剂量中以活性完整形式到达小肠的比例是药效的关键决定因素,38f的优异体内降糖效应,尤其是低剂量下的表现,可归因于其化学稳定性在上消化道恶劣环境中的战略性改善——HPLC稳定性研究表明,38f在模拟胃液(pH 1.3)中2小时后保留98.9%完整形式,而前期报道的类似物41在相同条件下发生大量酸催化水解(损失68.8%),41在小肠发挥抑制作用的活性物种可能包括原型药物及其活性显著降低的酸催化降解产物,38f在胃中的近定量存活确保了高口服活性,与酸不稳定的41在到达小肠靶酶前大量降解形成鲜明对比。
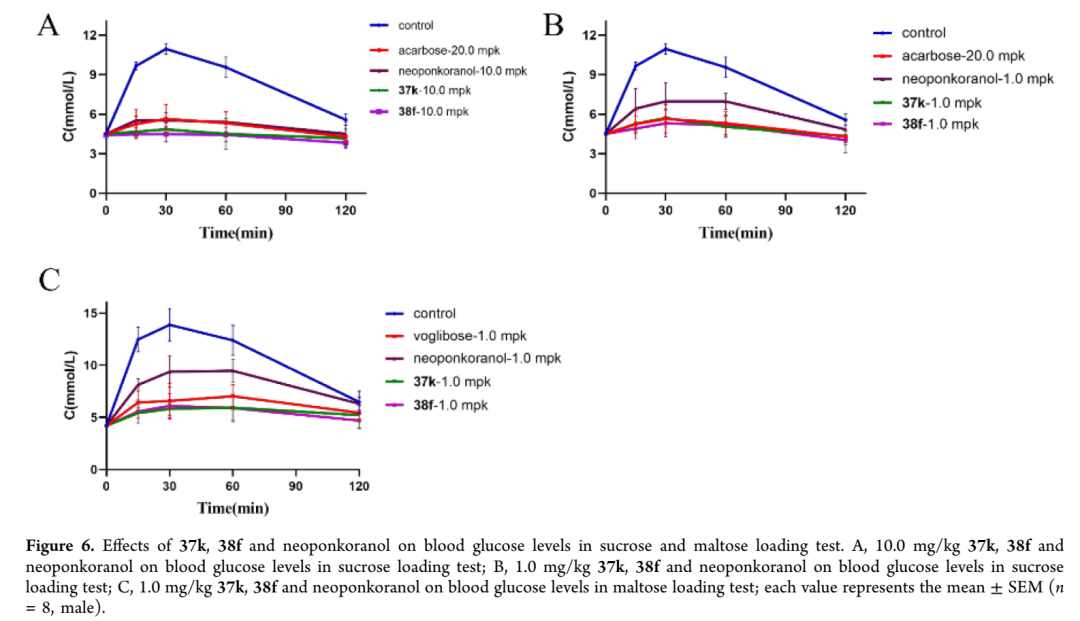
图片来源:ACS
⑤化合物对糖尿病小鼠空腹血糖、糖耐量及餐后血糖的影响
采用STZ诱导的糖尿病雄性小鼠评价38f的降血糖效果,设置高剂量(30 mg/kg)、中剂量(10 mg/kg)、低剂量(3 mg/kg)组,以阿卡波糖为阳性对照。每日灌胃给药后,通过每周空腹血糖(FBG)检测监测血糖状态。各药物均显示出降低糖尿病小鼠FBG的能力,模型组血糖较正常小鼠已升高3倍;给药28天后,30 mg/kg 38f组FBG较未治疗模型组降低46.8%,10 mg/kg组降低39.7%,3 mg/kg组降低32.8%,后者优于阿卡波糖50 mg/kg的24.3%降幅。口服葡萄糖耐量试验(OGTT)中,模型组呈现明显的餐后血糖激增且恢复延迟,38f显示清晰的剂量依赖性抗高血糖效应,3 mg/kg组仅轻度减弱血糖峰及AUC(降低37.7%),10 mg/kg组显著降低餐后峰值及AUC(降低46.5%),30 mg/kg组峰值抑制最强且60-120分钟曲线接近对照组、AUC降幅最大(49.5%),而阿卡波糖50 mg/kg组AUC降幅仅15.4%。胰岛素耐量试验(ITT)中,模型组对胰岛素的血糖降低反应迟钝,0-30分钟下降缓慢且60-120分钟仍维持高位,AUC最大,提示明显胰岛素抵抗;阿卡波糖作为机制特异性对照仅产生轻度改善,而38f呈剂量依赖性效应,3、10、30 mg/kg剂量相对于模型组在0-30分钟诱导血糖快速下降,30-60分钟更低,90-120分钟持续抑制,使整个曲线下移,AUC分析确认统计学显著改善且随剂量增强,10 mg/kg高度显著,30 mg/kg极高度显著,AUC值接近对照组,30 mg/kg未观察到过度低血糖证据,提示有效剂量范围内安全裕度充足。阿卡波糖和38f在ITT中改善胰岛素敏感性的效应,可能是持续降低餐后高血糖和糖毒性的继发性获益,38f的优越效应提示其更强的原发作用转化为更大的全身胰岛素抵抗辅助改善。餐后血糖试验采用麦芽糖和蔗糖负荷,二者均可被α-葡萄糖苷酶水解为葡萄糖,灌胃后的血糖水平间接反映α-葡萄糖苷酶活性。模型组灌胃麦芽糖后血糖持续显著升高,30分钟达峰,38f治疗显著减弱该升高,30 mg/kg组AUC较模型组降低48.2%,10 mg/kg和3 mg/kg组分别降低36.8%和27.2%,而阿卡波糖50 mg/kg仅降低22.2%,38f降低餐后血糖的优越性提示该锍盐候选物体内抑制α-葡萄糖苷酶活性更强。蔗糖耐量试验中获得相似结果,对照组15分钟血糖达峰后逐渐下降,而模型组和治疗组均30分钟达峰后下降,30 mg/kg 38f组蔗糖耐量改善最显著,120分钟血糖恢复正常至对照组水平,阿卡波糖组结果始终较差,甚至不及3 mg/kg 38f组;AUC分析显示38f 30、10、3 mg/kg剂量分别降低53.3%、46.3%、38.6%,均优于阿卡波糖50 mg/kg的28.4%。这些结果有力支持38f通过α-葡萄糖苷酶抑制改善体内麦芽糖和蔗糖耐量、抑制餐后高血糖。
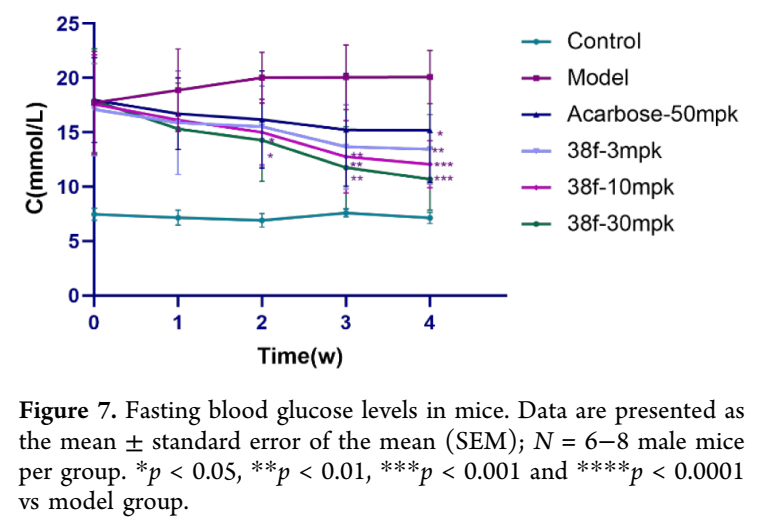
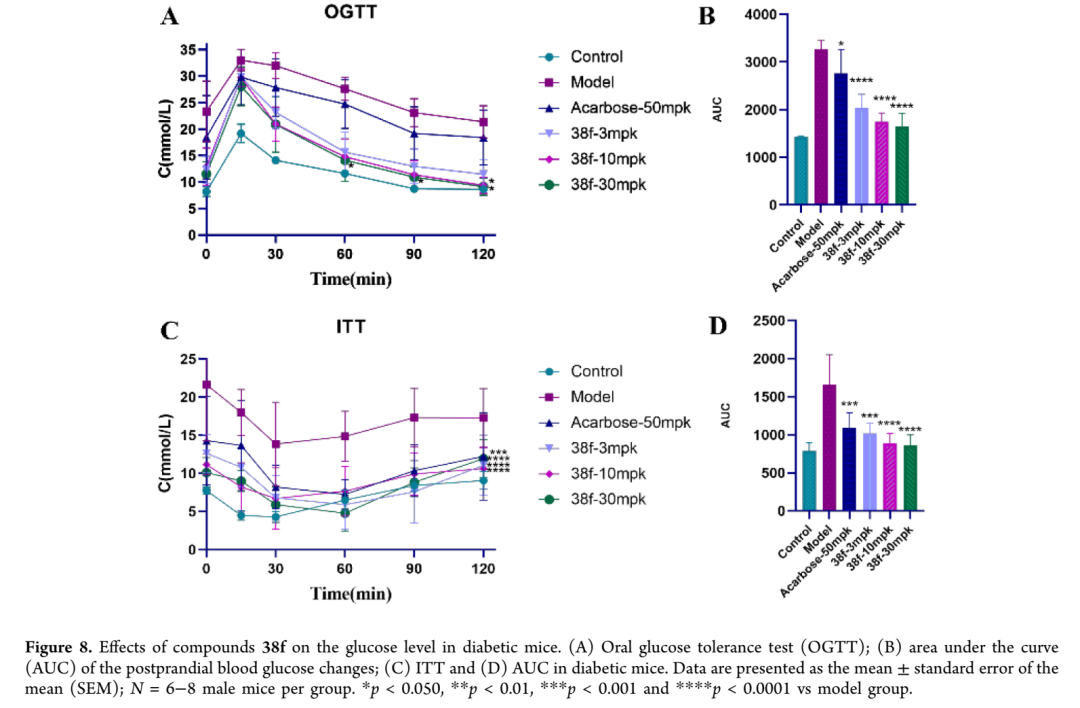
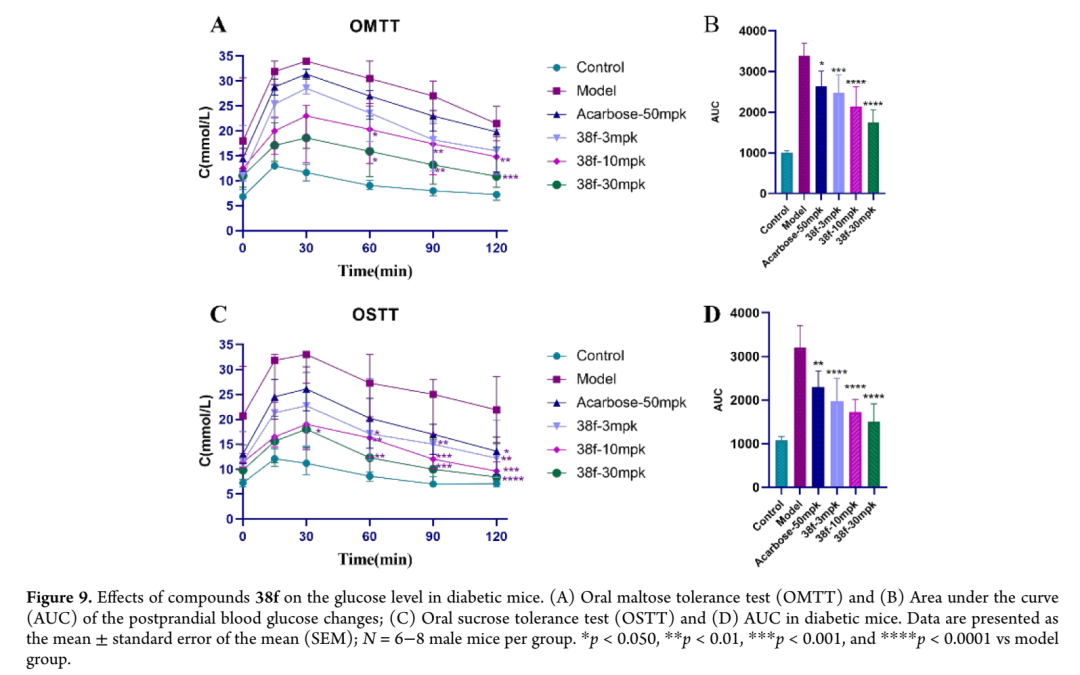
⑥对糖尿病雄性小鼠肝糖原和糖化血红蛋白(HbA1c)的影响
为评价化合物38f对糖尿病小鼠血糖控制的疗效,分析了肝糖原含量和糖化血红蛋白(HbA1c)水平。结果显示,糖尿病小鼠肝糖原储备显著降低,较对照组减少56.3%,可能归因于胰岛素缺乏和肝糖原合成受损;38f给药后呈剂量依赖性恢复肝糖原,30 mg/kg剂量在28天治疗后改善最显著。关于长期血糖控制,HbA1c作为不受短暂血糖波动影响的稳健生物标志物,是糖尿病诊断的关键参数;28天治疗后,38f呈剂量依赖性降低HbA1c水平,所有测试剂量均显示统计学显著疗效,其中30 mg/kg 38f的血糖控制效果优于阿卡波糖50 mg/kg,HbA1c降幅更大。肝糖原含量和HbA1c水平的剂量依赖性改善,有力表明38f通过增强血糖控制和改善肝脏碳水化合物代谢,有效缓解糖尿病相关代谢紊乱。
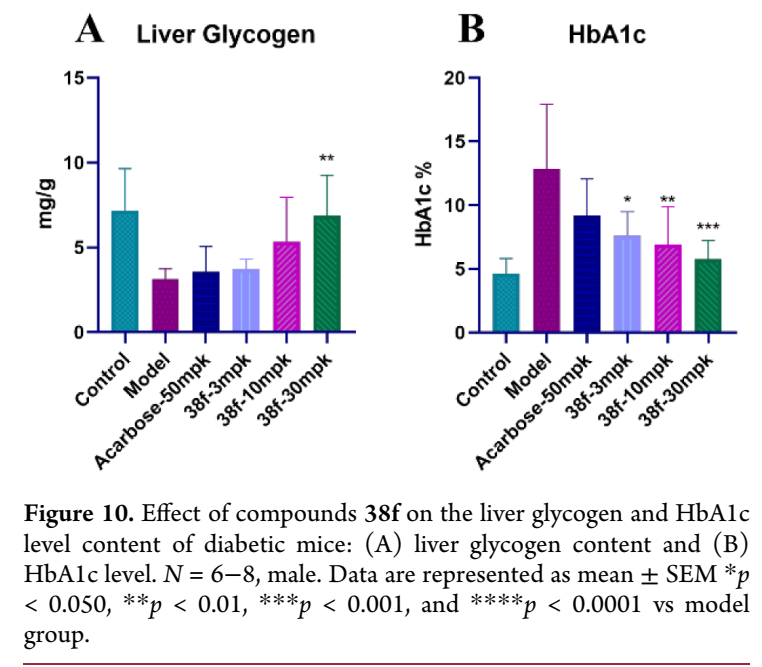
⑦38f的亚急性毒性评价
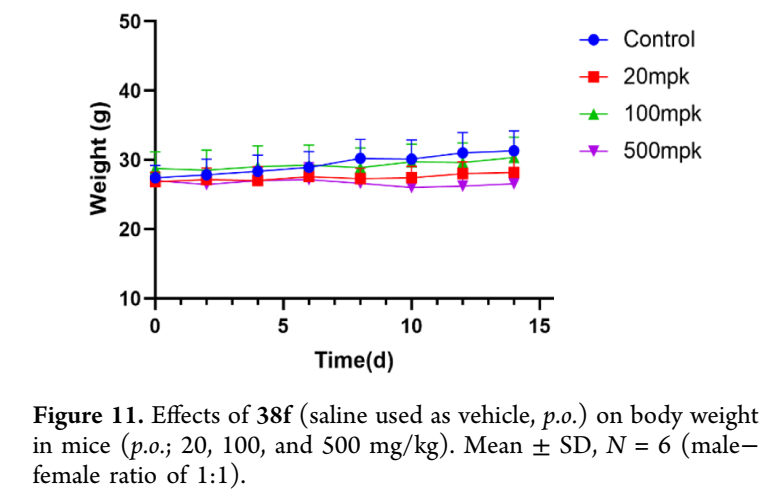
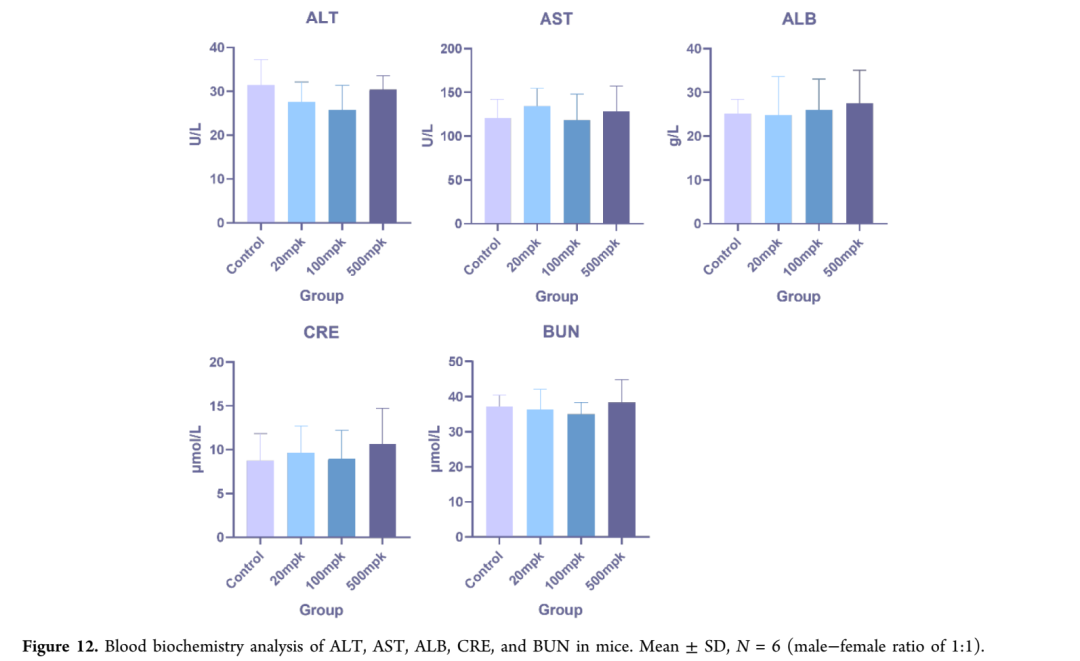
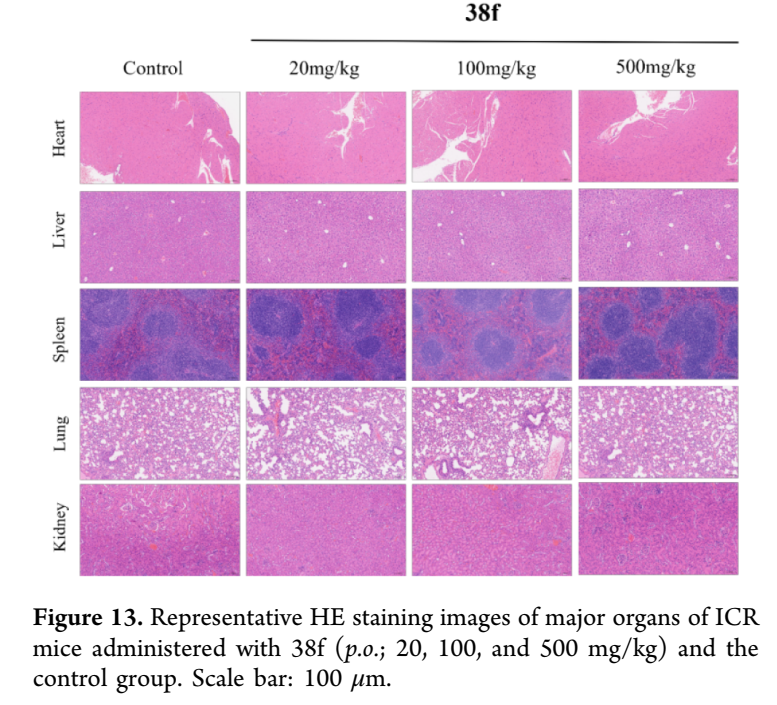
为进一步评价化合物38f的体内安全性特征,在ICR小鼠中进行了14天亚急性毒性研究,设置20、100和500 mg/kg三个剂量组。研究期间小鼠每日经灌胃口服38f,监测体重变化,未观察到异常行为。第14天处死小鼠进行尸检和组织采集,肝肾功能生化分析显示,肝功能标志物(天冬氨酸氨基转移酶AST、丙氨酸氨基转移酶ALT、白蛋白ALB)和肾功能参数(肌酐CRE、血尿素氮BUN)在38f治疗组与溶媒对照组之间无显著改变,无肝毒性或肾毒性证据。主要脏器(心、肝、脾、肺、肾)的脏器系数组间无统计学差异。苏木精-伊红(HE)染色组织病理学评价显示,各剂量38f处理小鼠的主要脏器均未观察到组织病理学改变。这些结果共同表明,38f即使在500 mg/kg剂量下仍表现出良好的安全性特征,无剂量依赖性毒性证据,结合其已证实的抗糖尿病疗效,提示38f适合进一步临床前开发和临床考察;在远超治疗浓度的剂量下(500 mg/kg)未显示显著毒性,提供了稳健的安全裕度,支持其作为糖尿病新型治疗候选药物的潜力。
图片来源:ACS
⑧化合物与ntMGAM及ctMGAM的分子对接研究
为阐明代表性配体的结合模式,对37k、37l、37m和38f与ntMGAM(PDB ID: 3L4U)进行对接分析。四个化合物均占据由酸性极性底部(Asp203/327/542/443)和疏水侧壁(Tyr299、Phe575、His600)组成的活性口袋,形成"极性锚定-疏水支撑"结合模式。所有化合物通过阳离子-π作用(5.7-6.1 Å)与Tyr299结合,并与Asp542、His600、Asp327、Asp203形成氢键。间位取代的37l无额外稳定作用,对接评分-6.854;对位三氟甲氧基取代的37m与Gln603形成卤键(2.4 Å),评分-6.950;邻位三氟甲基取代的37k额外形成氢键和π-π堆积,评分-7.078;2,5-二氯取代的38f形成双卤键(与Gln603 2.7 Å、Thr205 2.8 Å)及多点氢键网络,评分最优(-7.730)。值得注意的是,38f的锍阳离子未形成经典盐桥,而是通过阳离子-π作用与Tyr299结合,该模式在本系列化合物中普遍存在。硒原子较大的原子半径和更高极化率削弱阳离子-π作用,解释了锍盐普遍优于硒鎓盐的活性。与前期研究不同,38f未观察到π-π堆积,而是通过苯环双卤原子形成卤键双锚定,提示该作用模式可有效替代或补充π-π堆积。进一步将38f对接至ctMGAM(PDB: 3TOP),评分更优(-7.944),其通过氢键网络(Asp1157、Asp1279、Asp1526、His1584)、盐桥(Asp1420)、阳离子-π堆积(Trp1369)及疏水作用(Tyr1251)实现高亲和力结合。
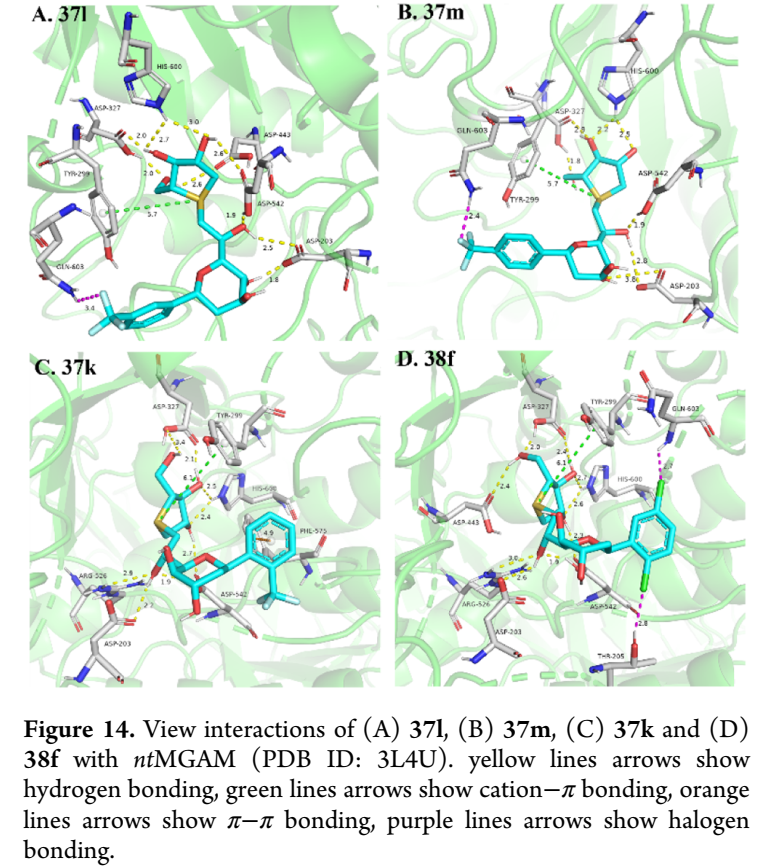
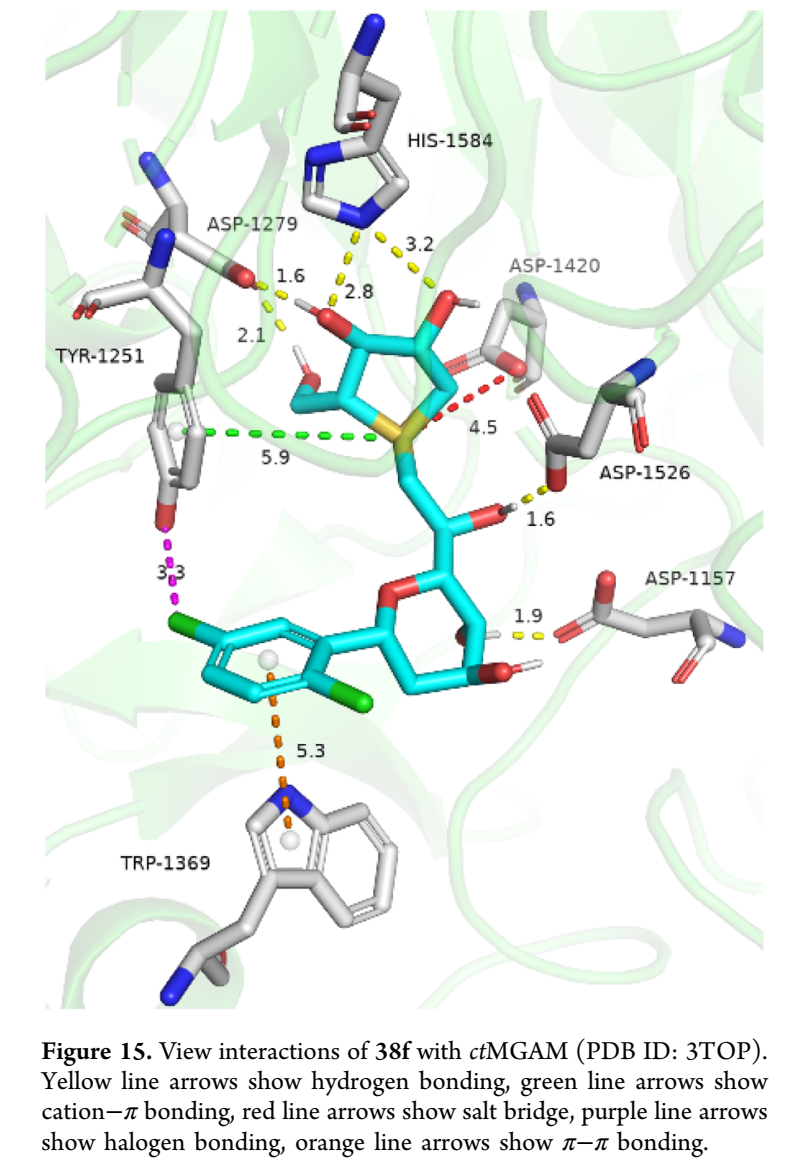
图片来源:ACS
总结与展望
通过理性药物设计、合成化学与综合生物学评价相结合的系统方法,研究者鉴定了一系列锍盐和硒鎓盐类候选α-葡萄糖苷酶抑制剂,其中化合物38f对关键碳水化合物消化酶蔗糖酶和麦芽酶表现出卓越的抑制活性。口服降糖药物开发的关键挑战之一是确保其在胃部恶劣酸性环境中的稳定性,基于前期构效关系研究的指导,具有特征性C-糖基吡喃侧链的锍盐38f在模拟胃液和肠液中均表现出优异的化学稳定性,提示高比例原型药物能以完整形式到达肠道作用部位。这种卓越的酸抗性不仅是理化性质的里程碑,更是增强药理性能的根本驱动力。在正常和糖尿病小鼠模型中,38f不仅有效降低餐后血糖,还改善糖耐量并降低糖化血红蛋白(HbA1c)水平,显示其长期血糖控制能力。糖尿病小鼠模型的全部生物学评价试验(包括OGTT、ITT、OMTT、OSTT、肝糖原和HbA1c)中,38f 3 mg/kg甚至表现出优于阿卡波糖50 mg/kg的显著治疗优势。在120分钟周期内,38f展现出强效降低血糖峰值并促进血糖恢复基线的能力,这种快速正常化现象结合慢性研究中空腹血糖和HbA1c的显著改善,为38f有助于整体血糖稳态恢复提供了有力的初步证据。亚急性毒性研究表明,38f即使在500 mg/kg剂量下仍表现出优异的安全性特征,未观察到显著肝毒性或肾毒性,结合其强效生物活性和稳定性,突显了该化合物作为安全有效T2DM治疗药物的潜力。本研究不仅深化了对强效α-葡萄糖苷酶抑制结构需求的理解,还获得了有前景的先导化合物38f,彰显了理性药物设计在解决未满足医疗需求中的力量,为开发靶向α-葡萄糖苷酶的下一代抗糖尿病药物铺平了道路。
文献详细全面信息请跳转原文阅读:
https://doi.org/10.1021/acs.jmedchem.5c02873
信息来源:药研视角




免责声明
“汇聚南药”公众号所转载文章来源于其他公众号平台,主要目的在于分享行业相关知识,传递当前最新资讯。图片、文章版权均属于原作者所有,如有侵权,请在留言栏及时告知,我们会在24小时内删除相关信息。
本平台不对转载文章的观点负责,文章所包含内容的准确性、可靠性或完整性提供任何明示暗示的保证。
喜欢的点个“看一看”和"喜欢"吧

不然微信推送规则改变,有可能每天都会错过我们哦~





