肿瘤基因变异检测试剂审评新规解读:边界更清晰,门槛再提升
发布时间:2026-03-27来源:基因谷
“更规范、更严格、更能拉开差距”时隔三个半月,CMDE发布了《肿瘤基因变异检测试剂技术审评要点(第二次征求意见稿)》。相比去年底的首次征求意见稿,在核心原则保持一致的情况下,对肿瘤NGS的试剂盒注册给出了更清晰的边界和门槛要求。“二次征求意见稿”相比之前有哪些变化?这些变化会带来哪些影响?
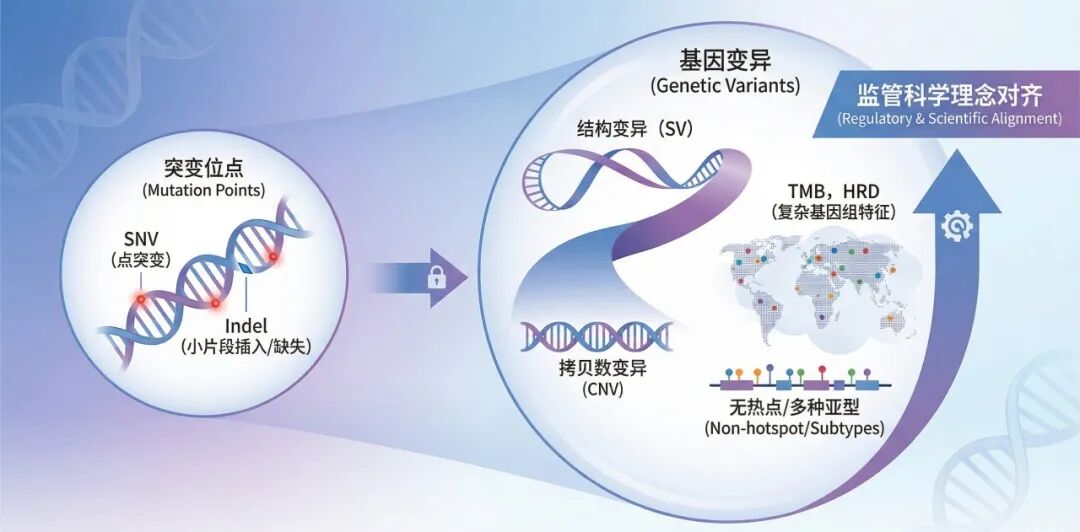
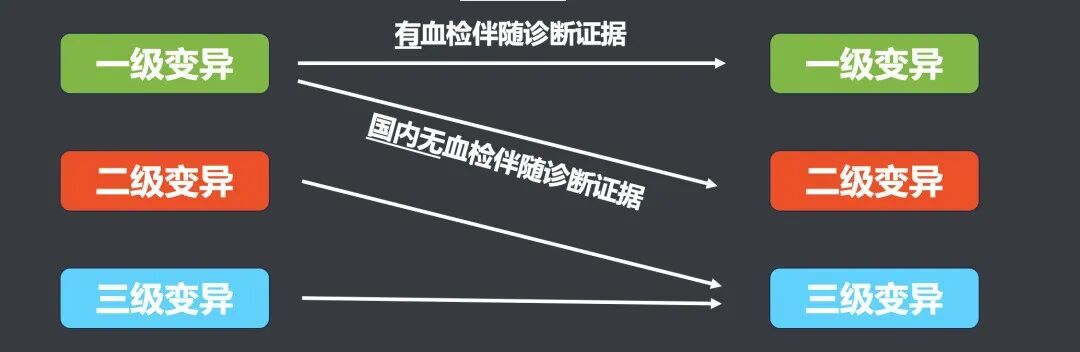
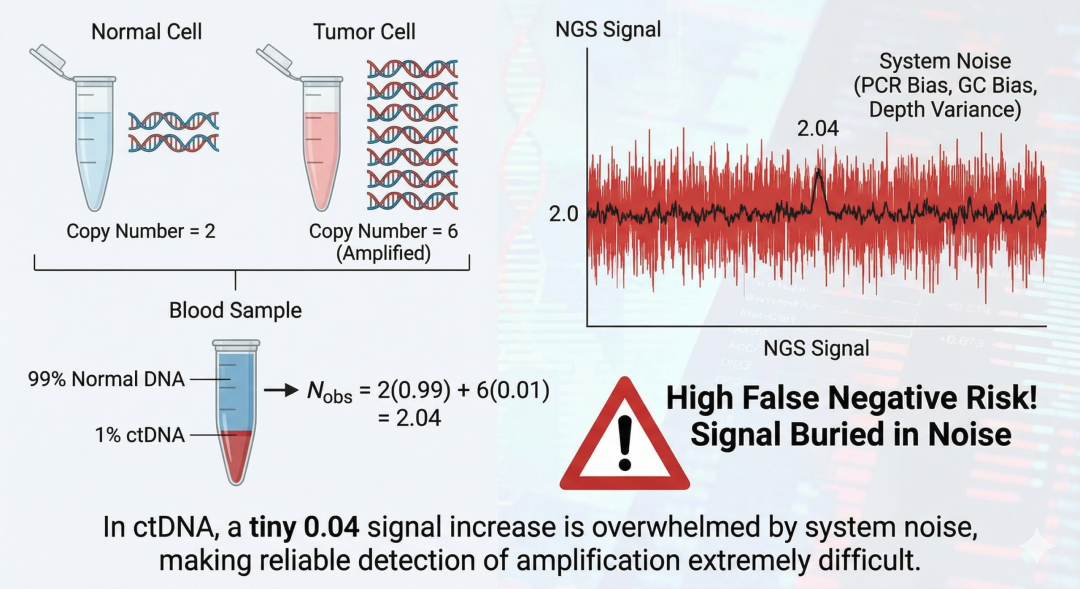
“基因变异”全面取代了“基因突变(位点)”既往的一级/二级/三级基因突变位点统一改成了一级/二级/三级基因变异。冷知识:“x级位点”这个叫法明确的出现在官方文件里是2023年9月份中国器审在公众号上发的《基于高通量测序技术(NGS)的肿瘤基因变异检测伴随诊断试剂的检测范围可以包括哪些基因及位点?》,随后被沿用至首次征求意见稿。相比于更容易被限定在点突变(SNV)、小片段插入/缺失(Indel)的“突变位点”,基因变异可以:这次从“突变位点”升级为“变异”,是监管层在科学概念和临床应用上向现代理念的全面对齐。二次征求意见稿没有改变“一/二/三级变异”这个框架,但对细节做了调整。二级变异的定义被缩窄了,部分原本被认定为二级的变异现在会被认定为三级。在新的征求意见稿中,下面这句话代表的变异被挪到了三级:“或者与抗肿瘤药物正在同步开发,已有研究数据初步显示其具有伴随诊断意义的基因突变位点”这意味着一个新的变异类型要想被注册为二级变异从而写进说明书,进入权威诊疗指南是唯一的路径。二级变异因为可以明确写进说明书并出具检测报告,是可以收费的。而收费的前提是“对临床有用”,一个没有指南推荐的同步开发中的变异,其“有效性”并不是那么稳固,毕竟药物的临床试验“中期获益、终点失败”的情景也不少见。不写进说明书,不可以出具报告,不能收费,但未来可以升级。另外,可能有朋友注意到血检的二级变异中加入了“在国内”这个限定词:但其实含义并没有变化,因为血检的二级变异对应组织检测的一级变异,而组织检测的一级变异原本就要求有中国人群的伴随诊断证据。二次征求意见稿中,血检二级变异的“禁区”新增了拷贝数变异:血检二级变异中建议慎重纳入基因融合、拷贝数变异和负向位点。当然,这不意外,因为拷贝数变异(CNV)在血检中一向是难点,因为ctDNA占比太低,所以敏感性相比组织检测差一些。但对于头部厂家来说,这也是证明技术领先性的绝佳机会。要想在血检产品中率先纳入这三个“禁区”,有且只有两条路:1. 去做原研CDx,拿到最直接的药效学证据,注册为一级变异2. 和肿瘤组织做同源对比研究,证明产品检测性能满足临床需求,注册为二级变异不管是哪条路径,率先完成这三个“禁区”在一级/二级变异注册的厂家,都可以很硬气的说:二次征求意见稿进一步强化了“原研CDx”的竞争优势。但在最容易被fast-follow的第四条路径中,加入了新的要求:(一致性比对)原则上应为境内已上市原研伴随诊断试剂。虽然“原则上”这个词看上去留有余地,但这个余地应该仅限于“境内无原研CDx”的情况,可以被理解成:如果境内已经有已上市原研伴随诊断试剂,就只能和它做一致性比对。仅当抗肿瘤药物开发中确实采用同一产品进行血浆和肿瘤组织同步检测,且该产品在产品设计上可以同时满足血浆检测和组织检测的需求时···二次征求意见稿则进一步明确了注册双样本IVD的门槛:双样本IVD中血检的基因变异必须是原研CDx,有最直接的药效学数据。想通过“一致性对比”去做双样本NGS产品中血检基因变异的注册,是不被允许的。二次征求意见稿中删掉了双样本IVD“仅限于小panel产品”的描述。这意味着拥有血检原研CDx合作的厂家有机会注册一个“大panel组织NGS+血检CDx变异”的大panel双样本产品。而没有血检CDx合作的厂家,将无缘双样本IVD产品。《肿瘤基因变异检测试剂技术审评要点(二次征求意见稿)》还有一些技术细节上的修改:比如三级位点的验证不再需要验证“每一套引物、探针结合区域”,比如一二级变异的临床验证增加了不同变异亚型的规范要求等等在保留核心原则的前提下,进一步提高了原研CDx的地位,是对行业走向“硬实力”竞争一个非常好的助力。对于“有新靶点原研伴随诊断的头部企业”是更大的利好和加强。


扫码加入基因谷行业交流群
转载说明:本文系转载内容,版权归原作者及原出处所有。转载目的在于传递更多行业信息,文章观点仅代表原作者本人,与本平台立场无关。若涉及作品版权问题,请原作者或相关权利人及时与本平台联系,我们将在第一时间核实后移除相关内容。
 五度妙笔
五度妙笔 企业透视镜
企业透视镜 API商城
API商城
 数据库
数据库