 五度妙笔
五度妙笔 企业透视镜
企业透视镜 API商城
API商城
 数据库
数据库【JMC】中国药科大学徐云根/邹毅:螺环喹啉类口服小分子TNF-α抑制剂的发现
2026.03.31 Online

近日,中国药科大学徐云根/邹毅团队在药物化学权威期刊JMC上发表了一篇题为"含螺环骨架的喹啉类口服小分子肿瘤坏死因子α(TNF-α)抑制剂的发现"的研究论文。
该研究针对类风湿关节炎(RA)治疗中生物制剂存在的感染风险高、肿瘤发生率增加及给药不便等临床痛点,基于骨架跃迁策略对AbbVie-CPD-12进行结构优化,设计并合成了一系列含喹啉骨架和螺环结构的小分子TNF-α抑制剂。其中,候选化合物XS-18展现出优异的TNF-α结合亲和力(FP IC₅₀ = 123 nM;Kᴅ = 45.9 nM),并在体外显著抑制TNF-α介导的炎症通路。在胶原诱导性关节炎(CIA)小鼠模型中,口服给药的XS-18表现出强效抗炎作用,其促进关节软骨修复的疗效优于临床药物托法替尼。尤为重要的是,XS-18具备出色的药代动力学性质(T₁/₂ = 7.1 h,口服生物利用度F = 56.8%)和良好的体内安全性,为开发口服小分子TNF-α抑制剂治疗自身免疫性疾病提供了具有临床转化潜力的新候选分子。
1min 速览
研究背景
类风湿关节炎(RA)是一种慢性进行性自身免疫性疾病,早期表现为关节肿胀疼痛,后期导致软骨破坏和骨侵蚀,最终造成关节畸形和功能障碍,严重影响患者生活质量。肿瘤坏死因子α(TNF-α)是RA治疗的重要靶点,目前临床使用的抗TNF-α生物制剂(如英夫利昔单抗、依那西普、阿达木单抗)虽疗效确切,但存在严重感染风险增加、肿瘤发生率升高及需注射给药等局限性。小分子TNF-α抑制剂具有口服给药便捷、生产成本低、严重副作用风险小等优势,且能选择性靶向可溶性TNF-α/TNFR1信号通路而不影响TNFR2介导的组织修复功能,因此开发新型口服小分子TNF-α抑制剂具有重要的临床价值。然而,目前仅有少数小分子TNF-α抑制剂进入临床试验,尚无该类药物获批上市,临床需求远未满足。
重点内容
研究团队基于AbbVie-CPD-12采用骨架跃迁策略进行理性药物设计,通过分子对接分析将BMS-CPD-42的喹啉骨架与优化后的螺环结构相结合,经多轮结构优化获得候选化合物XS-18;该化合物展现出优异的TNF-α结合亲和力(FP IC₅₀ = 123 nM,Kᴅ = 45.9 nM)和TNF-α/TNFR1相互作用抑制活性(IC₅₀ = 36.4 nM),在体外能有效抑制L929细胞凋亡、阻断NF-κB信号通路及炎症因子IL-6表达,且抗炎活性优于AbbVie-CPD-12,同时具备良好的药代动力学性质(口服生物利用度56.8%,半衰期7.1小时)和体内安全性;在CIA小鼠模型中,XS-18口服给药显著减轻关节肿胀、降低关节炎评分,其软骨修复效果优于临床药物托法替尼,并能有效下调踝关节组织中IL-6和TNF-α的表达,展现出作为口服小分子TNF-α抑制剂治疗类风湿关节炎的临床转化潜力。
研究总结
本研究通过骨架跃迁策略成功设计出新型喹啉类螺环小分子TNF-α抑制剂XS-18。该化合物具有以下突出特点:(1)优异的靶点结合亲和力,能直接结合TNF-α并破坏其三聚体对称性,选择性抑制sTNF-α/TNFR1相互作用;(2)显著的体内外抗炎活性,在细胞水平和CIA动物模型中均表现出优于参比化合物的抗炎效果,尤其在软骨修复方面优于临床药物托法替尼;(3)优良的药代动力学性质,口服生物利用度56.8%,半衰期7.1小时,支持口服给药;(4)良好的安全性,在急性和亚急性毒性研究中未观察到明显毒性反应。XS-18作为具有临床转化潜力的口服小分子TNF-α抑制剂,为类风湿关节炎等自身免疫性疾病的治疗提供了新的候选药物,有望克服现有生物制剂的局限性,为患者提供更安全有效的治疗选择。此外,该研究也为基于骨架跃迁策略开发蛋白-蛋白相互作用抑制剂提供了成功范例。
图片来源:ACS
详细阅读
01
INTRODUCTION
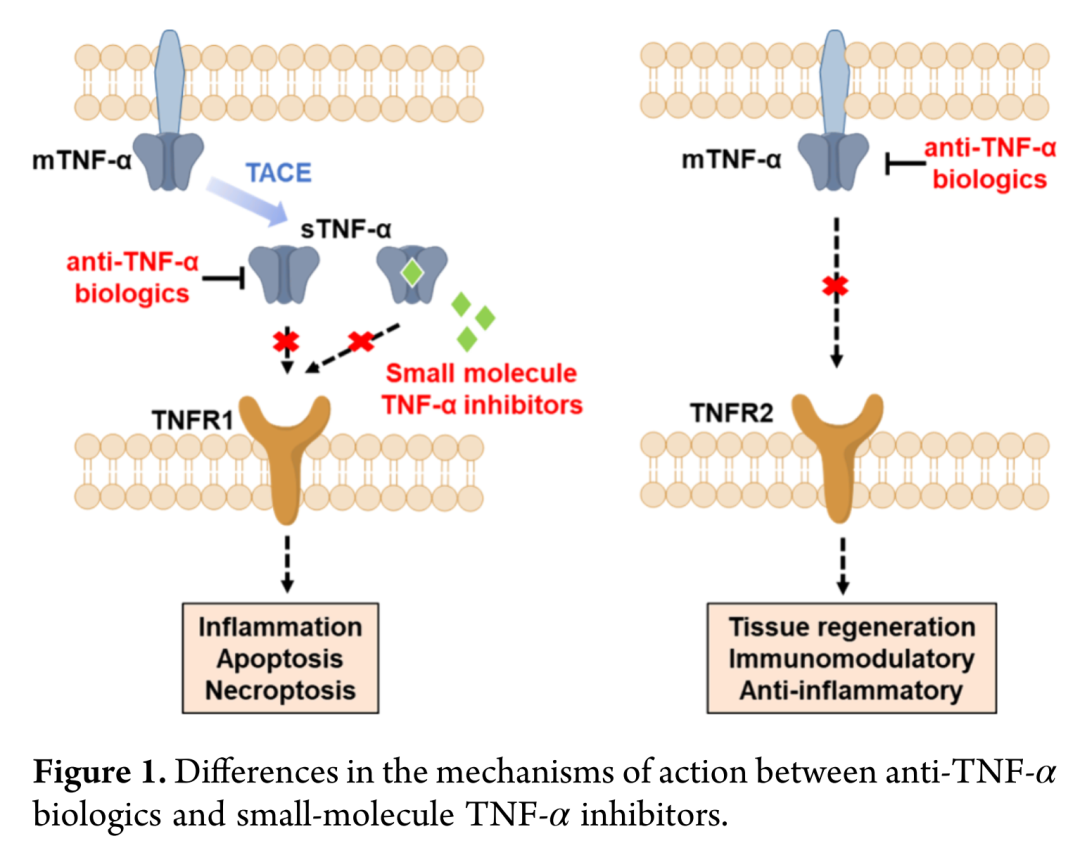
研究背景
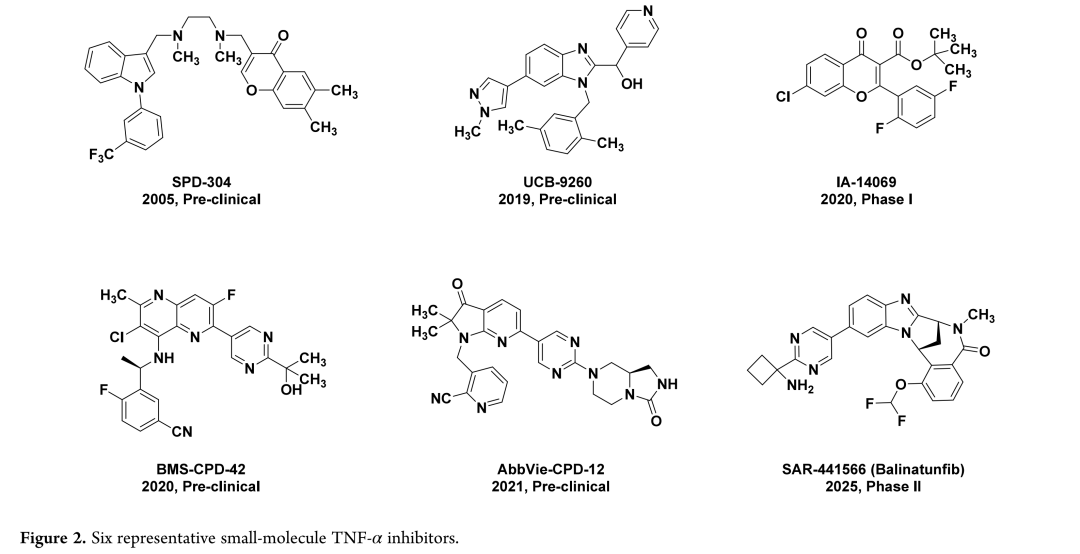
类风湿关节炎是一种慢性进行性自身免疫性疾病,早期表现为关节肿胀疼痛,后期导致软骨破坏和骨侵蚀,最终造成关节畸形和功能障碍;肿瘤坏死因子α在类风湿关节炎患者体内主要由M1型巨噬细胞分泌并介导炎症过程,靶向TNF-α是治疗该类疾病的成熟有效策略。目前已有多款抗TNF-α生物制剂获批临床,但存在严重感染和肿瘤风险增加等不良反应。TNF-α可分为跨膜型和分泌型两种形式,分别与TNFR1和TNFR2受体结合;抗TNF-α生物制剂对两种形式缺乏选择性,同时抑制TNFR1和TNFR2通路,而TNFR2通路介导组织修复,这种非选择性作用导致不良反应。相比之下,小分子TNF-α抑制剂选择性靶向分泌型TNF-α/TNFR1通路,具有口服方便、成本低、副作用小等优势。目前已发现六款代表性小分子抑制剂,仅两款进入临床试验,尚无药物获批,开发新型小分子TNF-α抑制剂意义重大。本研究基于AbbVie-CPD-12,通过骨架替换设计合成喹啉类衍生物,XS-18被鉴定为最有效候选化合物,在CIA模型中表现出显著抗炎疗效,有望成为口服治疗类风湿关节炎的候选药物。
02
RESULTS AND DISCUSSION
①基于骨架替换的理性设计
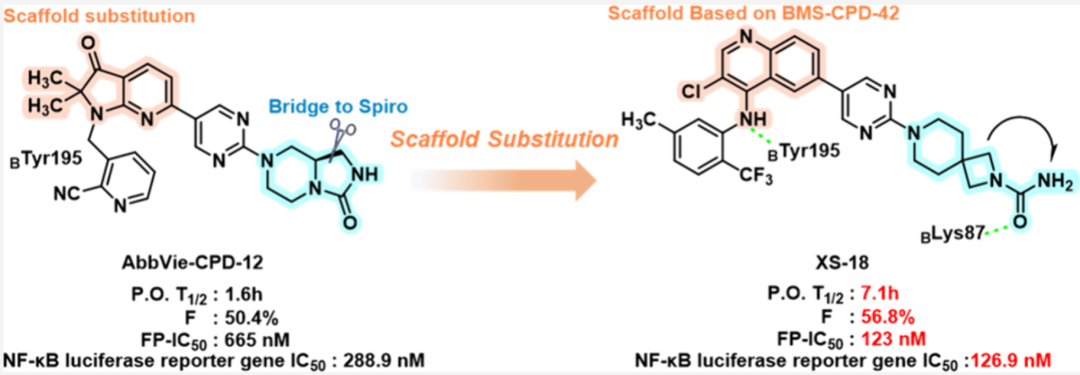
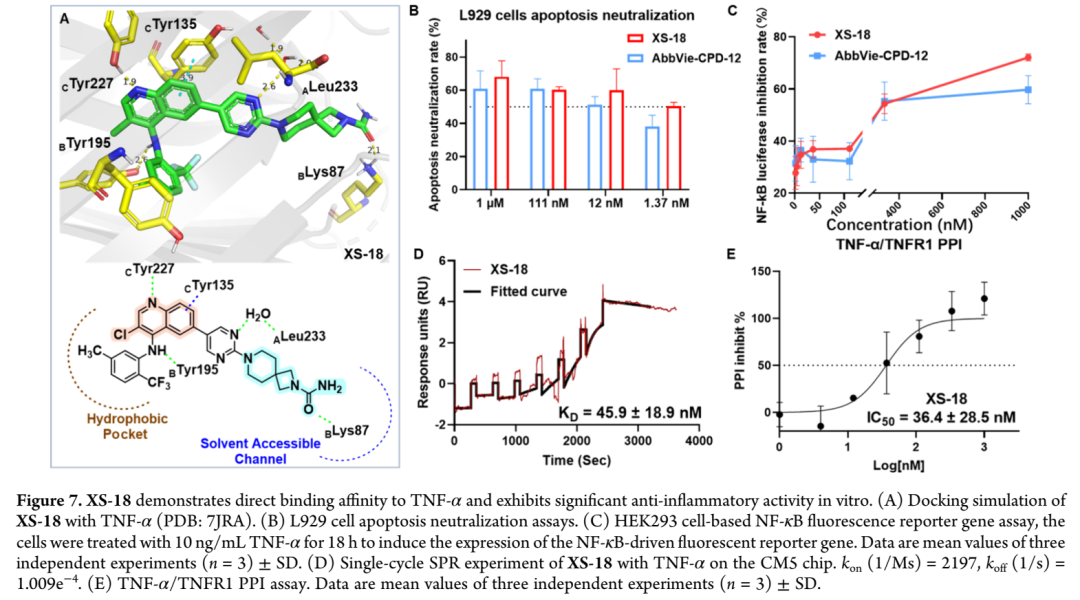
研究团队首先对AbbVie-CPD-12和BMS-CPD-42与TNF-α蛋白(PDB代码:7JRA)的预测结合模式进行了分析,以研究其不同的结构骨架的结合模式;结果表明,在TNF-α的疏水口袋中,AbbVie-CPD-12的1,2-二氢-3H-吡咯并[2,3-b]吡啶-3-酮骨架与CTyr227形成氢键相互作用,与CTyr135形成π-π堆积相互作用,而BMS-CPD-42的1,5-萘啶-4-胺骨架除上述相互作用外,还通过其-NH基团与BTyr195形成额外的氢键,苯胺基团与ATyr135形成π-π堆积相互作用;因此,与AbbVie-CPD-12相比,BMS-CPD-42在TNF-α结合口袋内参与更显著的分子相互作用,而AbbVie-CPD-12主要参与单一疏水相互作用。在溶剂可及通道中,AbbVie-CPD-12通过引入亲水性桥环结构比BMS-CPD-42的三级醇基团更好地占据该区域,但桥环未能与相邻极性氨基酸残基如BLys87形成关键相互作用。为解决这一局限,研究团队用BMS-CPD-42的骨架替换AbbVie-CPD-12的骨架以增强小分子与TNF-α在结合口袋内的相互作用,同时优化位于溶剂可及通道的桥环结构以促进与相邻极性氨基酸残基形成关键相互作用。
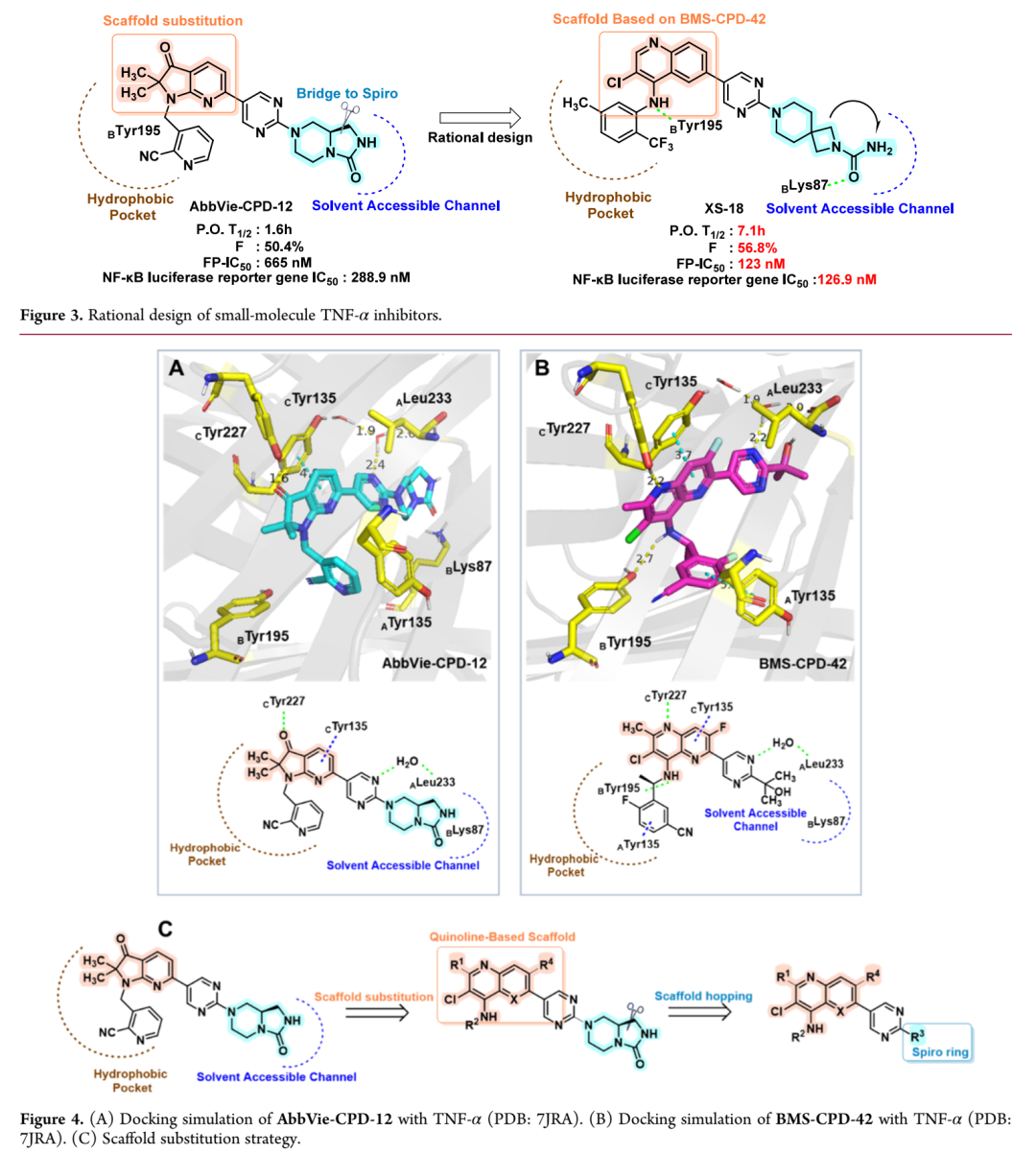
②基于结构的优化
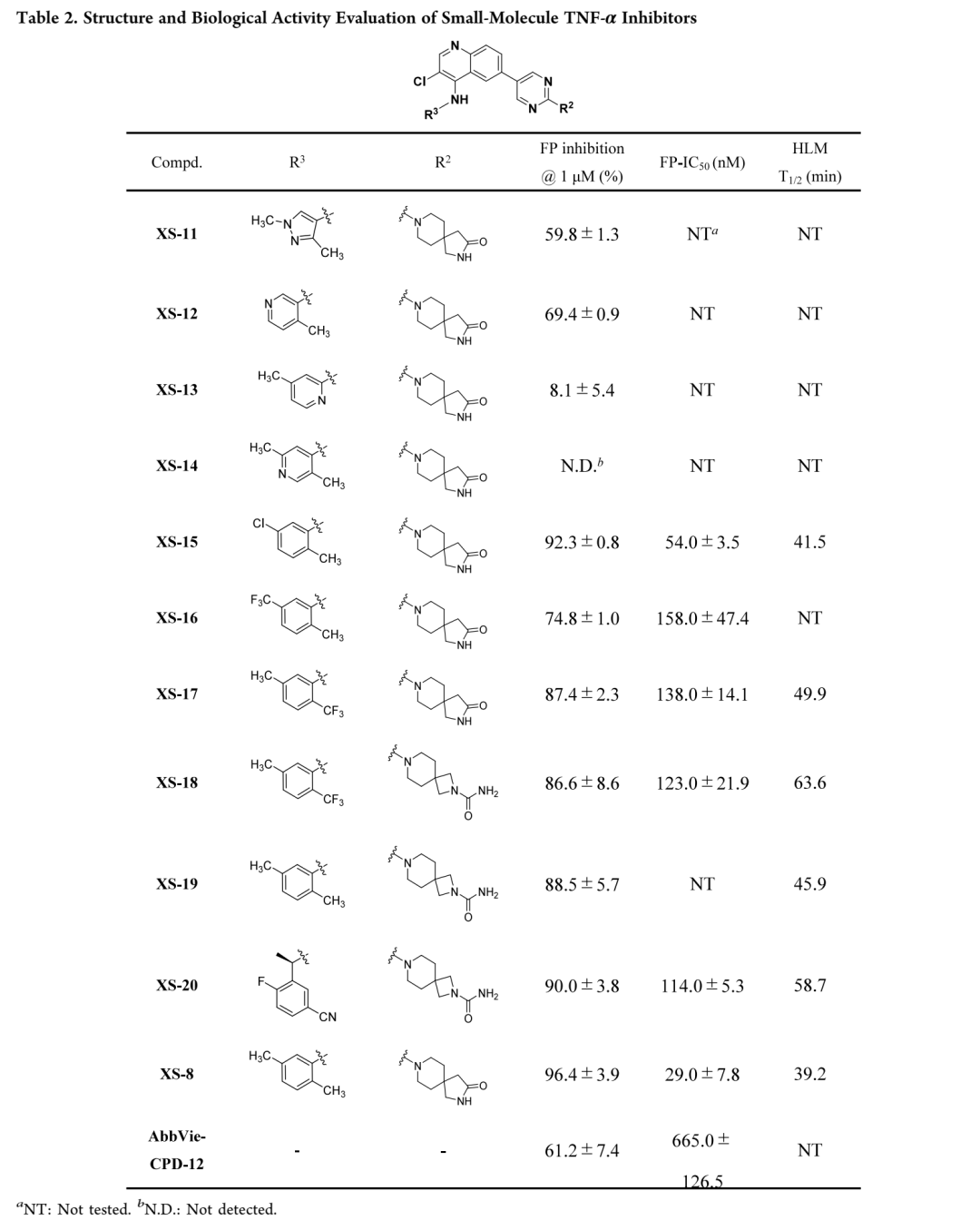
本研究采用荧光偏振(FP)结合实验筛选小分子与TNF-α的结合亲和力;针对AbbVie-CPD-12所用FP探针假阳性率高的问题,研究团队以喹啉为骨架重新设计了高亲和力FP探针。首先将AbbVie-CPD-12的吡咯并吡啶酮骨架替换为2-甲基喹啉,并通过引入带极性官能团的螺环结构修饰桥环体系;在4位引入2,5-二甲基苯胺基团的XS-1活性增强,验证了策略可行性。XS-2采用2,7-二氮杂螺[3.5]壬烷占据溶剂通道但活性较弱;将"-NH-"替换为羰基或"-CH(OH)-"得到XS-3和XS-4,亲和力显著增强,表明氢键供体更有利于结合。将氮杂环丁烷与嘧啶环偶联的XS-5因空间位阻增大导致活性降低。引入含螺环酰胺基团的XS-6和XS-7均优于AbbVie-CPD-12,但XS-7因失去与CAla232和BLys87的氢键相互作用而活性下降。将R1位"-CH3"替换为"-H"的XS-8结合亲和力进一步增强(FP IC₅₀ 29 nM),分子对接显示其消除了空间位阻,与BTyr195形成氢键并在溶剂通道形成更多相互作用。将"-NH-"替换为"-O-"的XS-9亲和力降低,证实该氢键的重要性。将喹啉替换为1,5-萘啶并引入氟原子的XS-10未改善亲和力,反而导致活性下降。
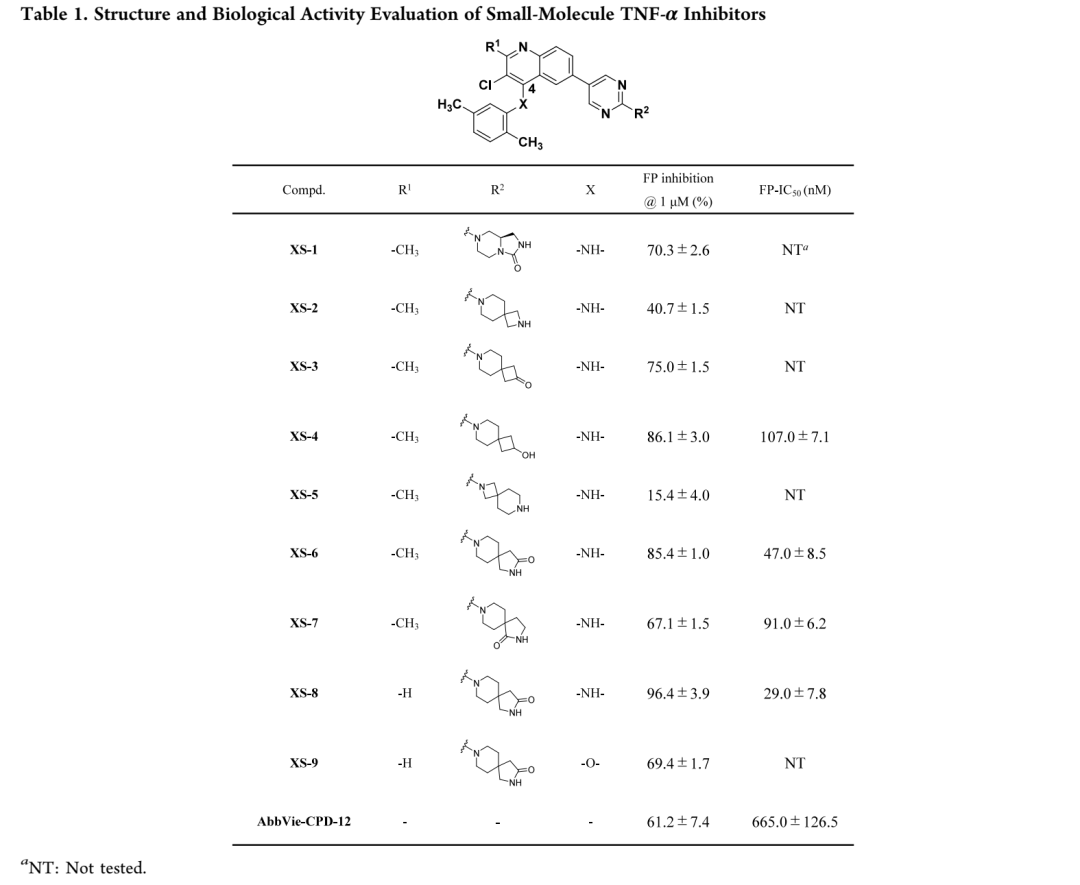
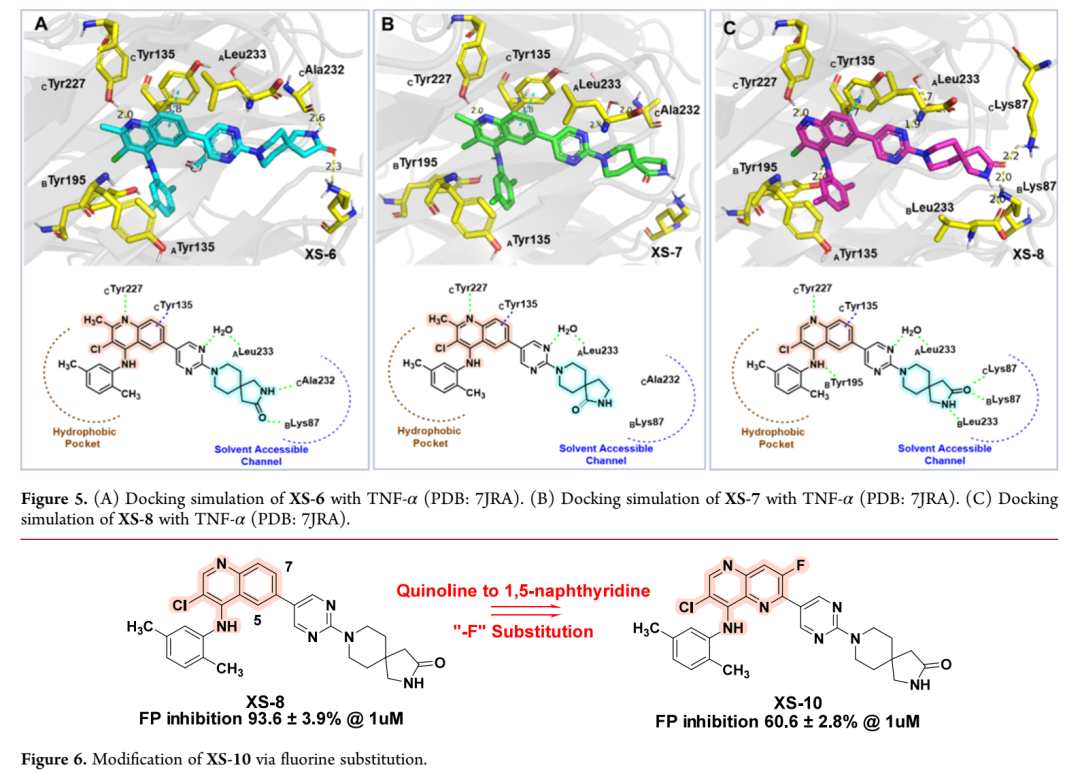
③体外代谢稳定性优化
体外人肝微粒体稳定性实验显示XS-8半衰期较短(T₁/₂ = 39.2 min),研究团队通过在2,5-二甲基苯胺骨架上引入吸电子基团进行优化。XS-11采用吡唑环替代但活性显著降低;XS-12和XS-13去除甲基导致亲和力下降,证实2位甲基的重要性;XS-14直接引入氮原子则几乎完全丧失活性。XS-15引入氯原子保持优异活性(FP IC₅₀ = 54 nM),XS-16和XS-17引入三氟甲基获得中等活性,半衰期分别延长至41.5 min和49.9 min;考虑到三氟甲基对口服生物利用度的改善作用,选择XS-17进一步优化。将五元螺环改为四元环、酰胺替换为脲基得到XS-18,代谢稳定性显著增强(T₁/₂ = 63.6 min)且保持抑制活性;XS-19验证脲基修饰确能提高代谢稳定性但活性略降,XS-20参考BMS-CPD-42改造后活性增强但代谢稳定性下降。鉴于XS-18良好的代谢特征和抑制活性,选择其进行后续体外抗炎活性评估。
④XS-18在体外显著抑制TNF-α介导的炎症通路
为评估XS-18与AbbVie-CPD-12的体外抗炎活性差异,研究团队进行了L929细胞凋亡中和实验和HEK293细胞NF-κB荧光素酶报告基因实验。在TNF-α(10 ng/mL)和放线菌素D存在下,L929细胞通过TNF-α/TNFR1/caspase-8信号通路发生凋亡,破坏TNF-α与TNFR1的相互作用可显著降低细胞凋亡率;1 μM浓度下,XS-18对L929细胞凋亡的抑制率为68.1±9.8%,同等条件下AbbVie-CPD-12为60.8±10.8%,且在1.37 nM的更低浓度下XS-18仍保持近50%的抑制效果,表明其效力优于AbbVie-CPD-12。TNF-α/TNFR1信号通路激活导致NF-κB核转位,促进下游炎症因子转录并加剧炎症反应,因此可通过检测稳定转染NF-κB报告基因的HEK293细胞中NF-κB依赖性荧光素酶活性的抑制效果来评估小分子TNF-α抑制剂的体外抗炎活性;首先评估了XS-18对NF-κB-TA-Luc HEK293细胞的细胞毒性,结果显示20 μM浓度下孵育72小时后细胞活力仍高于95%,表明XS-18对该细胞系毒性极小、生物相容性良好。荧光素酶实验显示,XS-18有效抑制NF-κB报告基因表达,IC₅₀值为126.9±33.8 nM,显著低于AbbVie-CPD-12(IC₅₀: 288.9±140.0 nM)。
图片来源:ACS
⑤XS-18抑制人类风湿关节炎成纤维细胞样滑膜细胞MH7A中IκBα磷酸化及IL-6表达
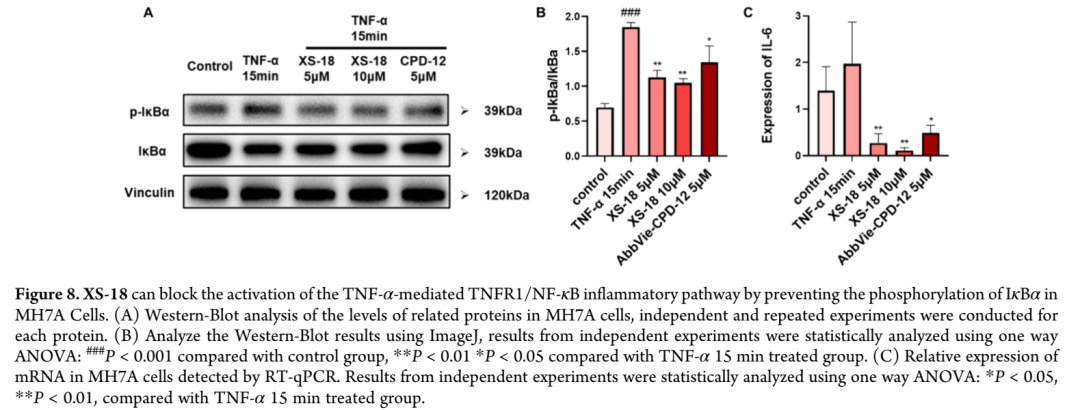
IκBα是TNF-α/TNFR1/NF-κB信号通路中的关键调控蛋白,该通路未被TNF-α激活时,IκBα可抑制NF-κB功能,阻止其转位入核;通路被TNF-α激活后,IκBα发生磷酸化、泛素化并随后降解,导致NF-κB释放并进入核内启动IL-6、IL-1β和TNF-α等靶基因的转录,促进炎症的发生发展。类风湿关节炎滑膜成纤维细胞作为关节炎症的主要参与者,可通过上述信号通路促进炎症进展,导致软骨组织损伤和关节功能障碍;基于此,研究团队选择人类风湿关节炎成纤维细胞样滑膜细胞MH7A,通过Western-Blot评估XS-18对TNF-α/TNFR1/NF-κB信号通路中IκBα磷酸化的抑制效果。结果显示,TNF-α(10 ng/mL)刺激MH7A细胞15分钟可显著上调p-IκBα/IκBα比值,5 μM和10 μM浓度的XS-18均能显著下调p-IκBα/IκBα,且5 μM浓度下对p-IκBα/IκBα的抑制效果比同等浓度的AbbVie-CPD-12更为明显,该结果更直接地证明XS-18可通过阻止IκBα磷酸化阻断TNF-α介导的TNFR1/NF-κB炎症通路激活,从而阻止NF-κB入核并转录相关促炎靶基因。实时定量PCR实验结果进一步表明XS-18如预期般下调IL-6表达,MH7A细胞本身表达IL-6,TNF-α(10 ng/mL)刺激15分钟后IL-6表达上调,5 μM和10 μM浓度的XS-18均能显著抑制IL-6表达,且效果优于AbbVie-CPD-12,表明XS-18具有更优的抗炎性质。
⑥XS-18的体内药代动力学及安全性评价
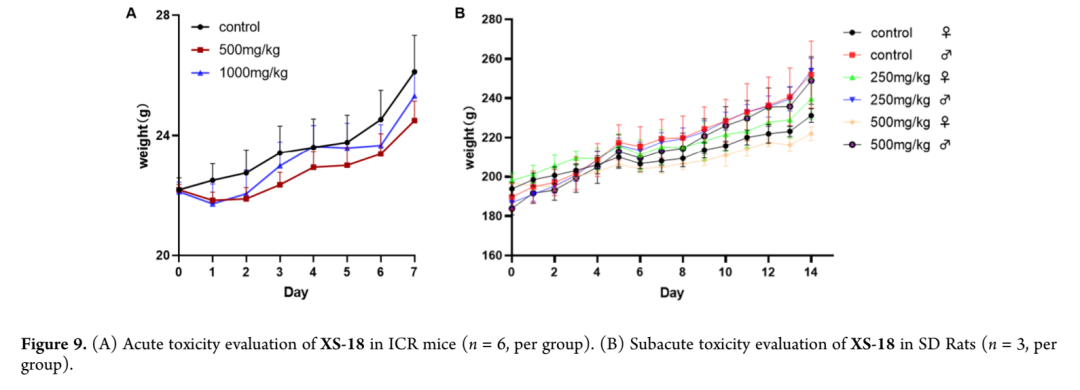
研究团队在大鼠中评估了XS-18的药代动力学性质,结果显示XS-18具有优异的口服生物利用度(F = 56.8%)和较长的半衰期(T₁/₂ = 7.1 h),AUC₀₋∞表现良好,提示其作为口服小分子TNF-α抑制剂的潜力。基于药代动力学数据,研究团队进一步通过小鼠急性毒性实验和大鼠亚急性毒性实验评估了XS-18的体内安全性:在ICR小鼠急性毒性实验中,单次口服给药500 mg/kg或1000 mg/kg后7天观察期内未观察到显著体重下降或异常行为改变;在SD大鼠亚急性毒性实验中,连续14天每日口服给药250 mg/kg和500 mg/kg后,各处理组与对照组相比体重无显著下降,心、肝、脾、肺、肾等主要脏器指数分析显示各组间脏器/体重比值无统计学差异,表明实验条件下XS-18未引起可检测的器官毒性。

⑦XS-18体内抗炎疗效评价
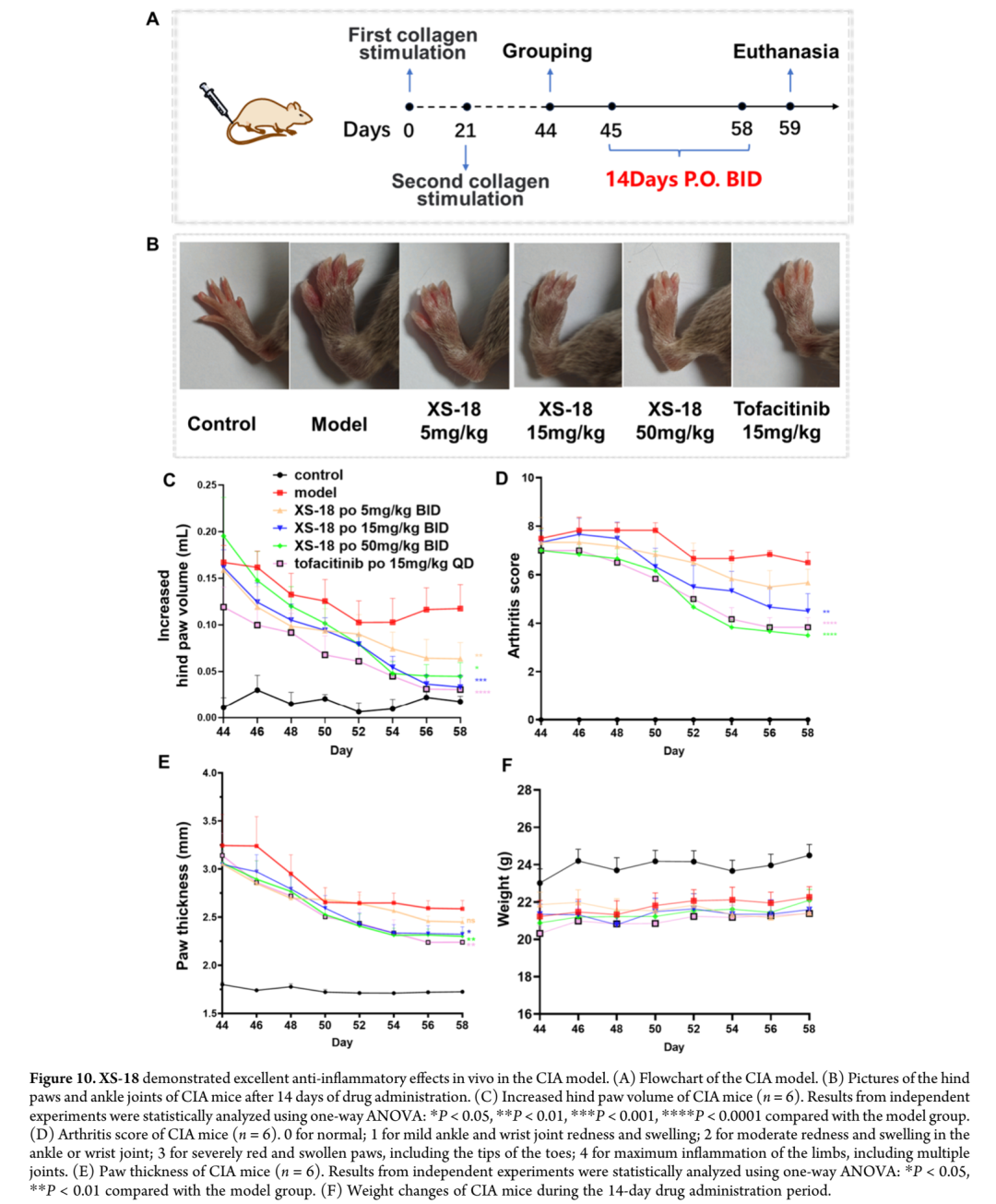
研究团队采用胶原诱导的类风湿关节炎小鼠模型(CIA)评估XS-18的体内疗效,并与已获批用于治疗类风湿关节炎的首个口服靶向合成改善病情抗风湿药(tsDMARD)托法替尼进行比较;托法替尼作为泛JAK抑制剂,与主要心血管事件和恶性肿瘤风险增加相关。DBA/1J小鼠经两次胶原刺激成功建立类风湿关节炎模型,基于XS-18的药代动力学特征,选择5、15和50 mg/kg三个剂量水平,连续14天每日两次口服给药,监测后足体积平均增加量;前8天(第44-52天)模型组小鼠后足肿胀呈下降趋势,提示存在自发愈合活性,但第9至14天(第52-58天)后足体积增加速率开始上升,表明自愈能力已达平台期。实验中,XS-18中剂量组(15 mg/kg)对小鼠后足体积增加的抑制效果最为显著,治疗14天后与托法替尼组相当,值得注意的是托法替尼组治疗开始时后足体积增加基线水平最低;XS-18高剂量组(50 mg/kg)也表现出良好的抑制效果,但由于初始基线较高,14天后疗效略低于中剂量组;低剂量(5 mg/kg)和中剂量(15 mg/kg)组基线水平相近,低剂量对后足体积增加的抑制效果相对较弱。除后足体积增加外,爪厚度变化也是评估类风湿关节炎进展的关键参数,由于治疗开始时各组基线测量值相近,XS-18对爪厚度的抑制作用呈剂量依赖性;高剂量组(50 mg/kg)爪厚度显著降低,与托法替尼组效果相当,中剂量组(15 mg/kg)也表现出统计学显著的抑制效果。为更全面评估小鼠类风湿关节炎的发生发展,包括踝关节和足爪红肿程度等临床指标,对各药物治疗组小鼠关节状况进行评估以确定关节炎评分;高剂量组(50 mg/kg)关节炎评分显著降低,优于托法替尼治疗组,中剂量组表现出中度降低,低剂量组无明显治疗效果。治疗结束时,踝关节和足爪的摄影记录直观证实了XS-18高剂量组的显著抗炎效果,红肿明显缓解,改善程度超过托法替尼组;给药期间各组体重无显著变化,主要器官H&E染色未见明显组织病理学异常,支持口服XS-18的良好安全性特征。
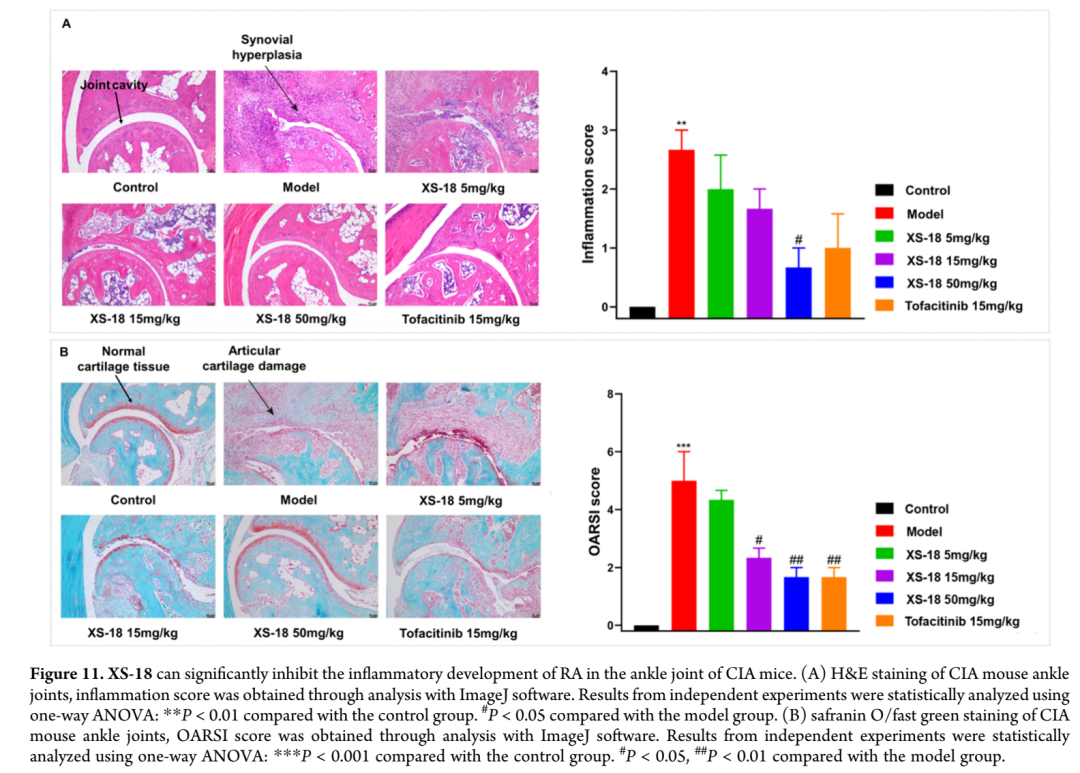
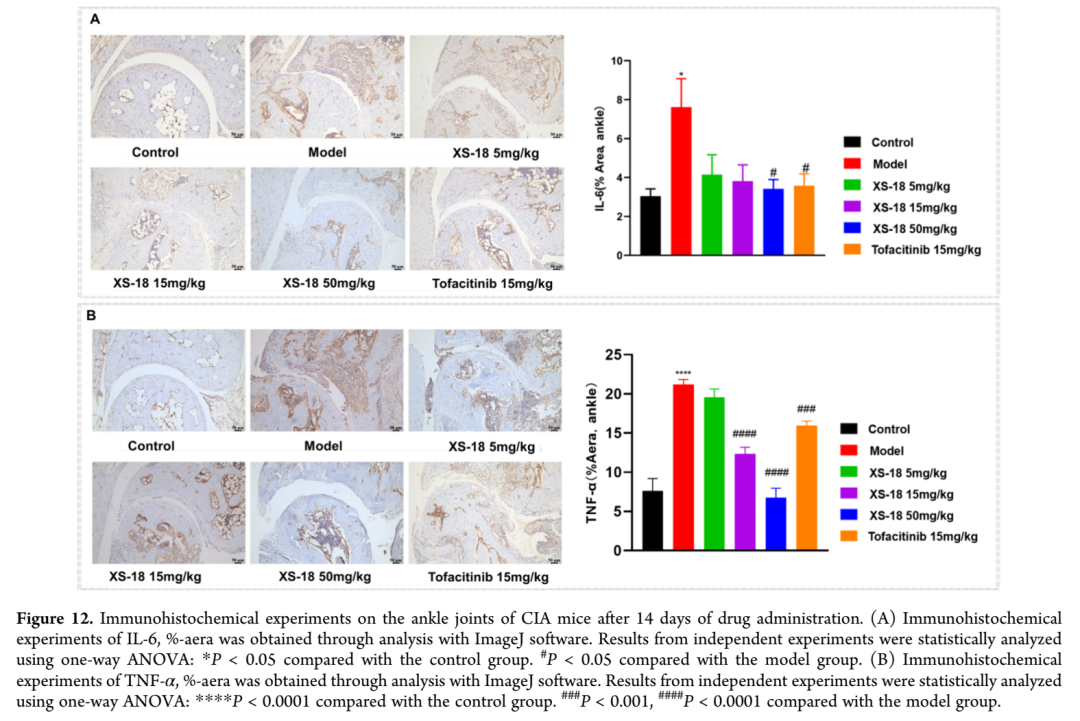
图片来源:ACS
总结与展望
综上所述,研究团队通过骨架替换策略成功设计并合成了XS-18,这是一种源于先前报道的AbbVie-CPD-12的新型TNF-α小分子抑制剂;XS-18在FP和SPR实验中表现出对TNF-α的强结合亲和力,并显示出显著的体外抗炎活性。此外,XS-18具有良好的体内安全性特征和稳健的药代动力学性质(T₁/₂ = 7.1 h,F = 56.8%),提示其作为口服TNF-α抑制剂的良好潜力。在CIA模型中,XS-18通过有效抑制踝关节组织中IL-6和TNF-α的表达水平表现出显著抗炎活性,其治疗效果超过托法替尼。这些发现表明,作为小分子TNF-α抑制剂,XS-18有望成为一种有效的口服tsDMARD,可能为托法替尼提供替代选择,同时降低严重临床不良反应的风险。鉴于自身免疫性疾病潜在的复杂病理机制,除TNF-α外,TACE、受体相互作用蛋白激酶1/3(RIP1/3)和IL-17A等其他蛋白在炎症发展中也发挥关键作用,因此开发调节TNF-α与其他炎症相关蛋白活性的双靶点抑制剂有望实现对自身免疫性疾病的更全面调控。
文献详细全面信息请跳转原文阅读:
https://doi.org/10.1021/acs.jmedchem.5c03457
信息来源:药研视角




免责声明
“汇聚南药”公众号所转载文章来源于其他公众号平台,主要目的在于分享行业相关知识,传递当前最新资讯。图片、文章版权均属于原作者所有,如有侵权,请在留言栏及时告知,我们会在24小时内删除相关信息。
本平台不对转载文章的观点负责,文章所包含内容的准确性、可靠性或完整性提供任何明示暗示的保证。
喜欢的点个“看一看”和"喜欢"吧

不然微信推送规则改变,有可能每天都会错过我们哦~





