口服多肽可开发性评估:胰岛素与司美格鲁肽们的异轨殊途
发布时间:2026-04-14来源:药事纵横
口服多肽药物的开发长期以来被视为药物递送领域的“圣杯”。相较于传统的注射给药,口服途径具有患者依从性高、无需专业医疗人员辅助、适合慢性病长期治疗等显著优势。然而,多肽药物固有的理化性质与胃肠道极端的生理环境之间存在着巨大的冲突,导致口服生物利用度极低,通常不足1%,甚至低于0.1%。这一困境构成了口服多肽可开发性评估的核心命题。本文将从分子基础、生理屏障、设计策略、药代动力学特征、制剂技术以及临床转化经验等多个维度,系统阐述口服多肽药物的可开发性评估框架。1.口服多肽开发的科学基础与生理屏障
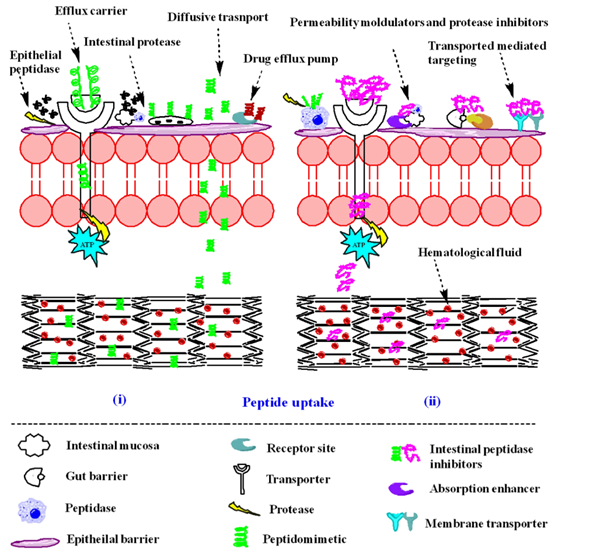
口服多肽药物的成功开发首先取决于对其分子特性与生理屏障之间相互作用的深刻理解。多肽通常由不超过50个氨基酸组成,分子量低于5000 Da,介于传统小分子和高分子量生物制剂之间。(含有51个氨基酸、分子量为5.8 kDa的胰岛素通常被视为分界线,既可称为多肽也可称为蛋白质。)这类分子具有优异的选择性、高效能和低毒性等内在优势,能够精准调控生物通路,在糖尿病、癌症、自身免疫疾病等多个治疗领域展现出广阔前景。然而,正是这些有益的分子特性,同时也带来了口服给药的固有局限。从理化性质来看,多肽药物的分子量通常超过500 Da,且随着氨基酸数量增加,亲水性显著增强。这种高亲水性导致多肽在脂质双分子层中的溶解度极低,难以通过跨细胞途径被动扩散。多肽在口服途径中面临的是一场“生存竞赛”,必须依次通过化学屏障、酶屏障和物理屏障才能进入体循环。胃肠道环境是一个严酷的化学工厂:胃液的强酸性可能导致多肽的化学降解或构象改变,而肠液的碱性环境同样构成挑战。更为致命的是酶屏障:胃中的胃蛋白酶、小肠中的胰蛋白酶、糜蛋白酶、羧肽酶等构成了强大的水解体系,多肽分子中的肽键是这些酶的特异性底物,导致多肽在到达吸收部位前往往已被降解为无活性的片段。这种“首过代谢”效应极大地降低了药物的生物利用度。图1 (i)肠道黏膜屏障的示意图,突出其对肽药物口服递送所面临的挑战。(ii)绕过肠道屏障以改善药物吸收的关键策略概述肠上皮表面覆盖的黏液层构成了另一道物理屏障。黏液层主要由黏蛋白组成,形成动态的网状结构。黏蛋白网状结构孔径较小,且带有负电荷,能有效截留亲水性大分子和纳米颗粒。多肽药物若要穿透上皮细胞,必须首先穿透这层黏液屏障。最后一道关卡是肠上皮屏障,肠上皮细胞通过紧密连接形成选择性屏障,紧密连接通常仅允许小分子通过,对于大分子多肽几乎关闭。尽管某些渗透促进剂可以可逆地打开紧密连接,但这同时也可能带来安全隐患。2.分子设计策略:突破稳定性与渗透性瓶颈
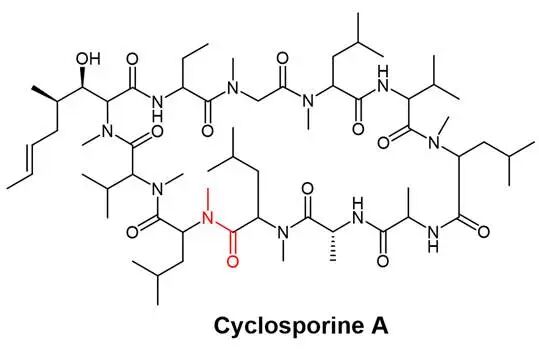
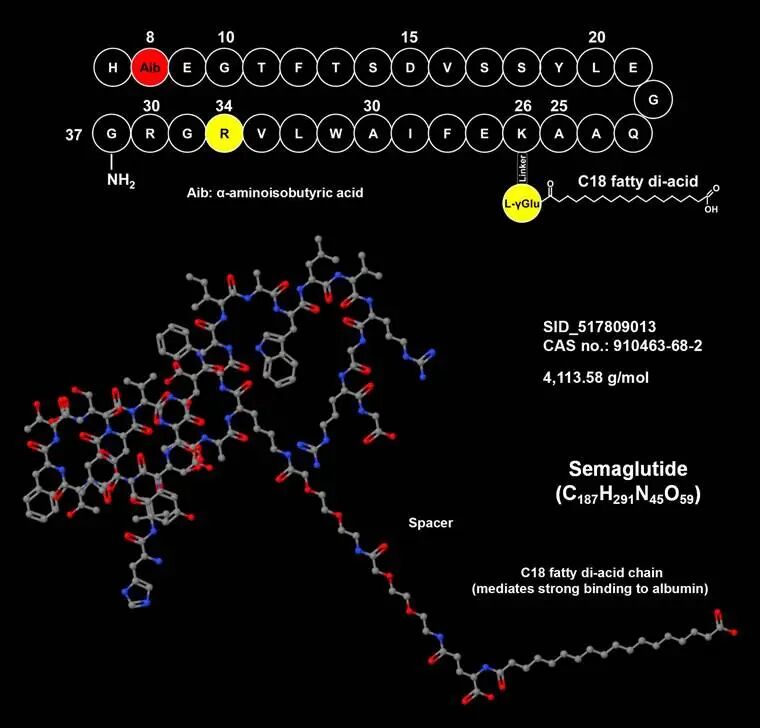
针对上述生理屏障,分子修饰是提升多肽口服可开发性的根本手段。通过对多肽分子结构的精准改造,可以优化其稳定性、渗透性和药代动力学特性。目前主要有三种修饰策略被广泛应用:氨基酸修饰、引入非天然氨基酸、以及脂质或聚乙二醇链。图2 环孢素A的化学结构环化是提升多肽口服适应性的核心技术之一。通过形成环状结构,可以显著改变多肽的物理化学性质:环肽的环化结构减少了自由端,显著降低蛋白酶切割位点,使其对胃酸和蛋白酶具备更高的抵抗力,显著提高了代谢稳定性;同时,环化限制了多肽的构象自由度,使其预先形成适合受体结合的构象,并降低极性表面积,有助于改善膜渗透性。环化策略包括头-尾连接、头-侧链连接、侧链-侧链连接等多种方式。口服环孢素A的成功应用,充分证明了环化策略的可行性。引入非天然氨基酸是增强酶稳定性的经典策略。蛋白酶通常特异性识别L-构型氨基酸,替换为D-构型可显著抵抗酶解。主链氮上的甲基化修饰不仅能阻断氢键供体、降低极性表面积,还能显著提高多肽的膜渗透性,并抑制蛋白酶的水解作用。这一策略在环状肽的设计中尤为关键,常被称为“N-甲基化扫描”以寻找最佳渗透性位点。聚乙二醇化是将聚乙二醇链共价连接到多肽分子上的策略,通过增加多肽的流体动力学体积,显著降低肾脏清除率,从而延长体内半衰期。聚乙二醇链在多肽表面形成的“保护伞”结构,能够屏蔽蛋白酶的攻击位点,抑制酶介导的降解。然而,聚乙二醇化也可能导致多肽的生物活性下降,且分子量的显著增加可能进一步阻碍跨膜转运,因此需在半衰期延长与渗透性之间寻找平衡。脂质化修饰通过脂肪酸链修饰增加白蛋白结合率,进一步延长半衰期并改善渗透性。司美格鲁肽的成功应用充分证明了这一策略的潜力。前药策略则是在多肽分子上连接特定基团,使其在胃肠道环境中暂时“失活”或“隐身”,以抵抗酶解或提高渗透性,待吸收进入体循环后,在体内特定环境下释放原药。图3 司美格鲁肽化学结构3.制剂技术与给药系统的创新突破
分子修饰解决了部分“内因”问题,但单纯依靠分子改造往往不足以达到临床所需的生物利用度。制剂技术和给药装置的创新则从“外因”层面提供了关键支持。渗透促进剂是目前最传统且应用最成功的策略之一。这类辅料能可逆地破坏肠上皮细胞的紧密连接或增加细胞膜流动性,促进多肽通过旁细胞途径或跨细胞途径吸收。司美格鲁肽口服制剂的成功应用了SNAC作为渗透促进剂,SNAC不仅提高药物局部浓度,还能短暂改变上皮细胞膜流动性,促进跨细胞转运。癸酸钠是研究最广泛的渗透促进剂之一,但其临床转化受到胃酸pH影响的挑战。研究表明,犬类和人类胃pH的差异可能导致癸酸钠制剂在不同物种间的表现差异,通过在制剂中添加pH调节剂可部分缓解这一问题。然而值得注意的是,即使与渗透促进剂联用,多肽的口服生物利用度仍通常仅为低个位数百分比。长期使用渗透促进剂可能破坏肠道屏障功能,引发炎症或增加毒素吸收风险,因此对其机制的深入研究仍在持续。纳米技术为多肽提供了“保护伞”和“运输车”。将多肽包载于脂质体或聚合物纳米粒中,可以有效隔绝酶环境,并利用表面修饰增强黏液粘附性或穿透能力,改善细胞摄取效率。自乳化药物递送系统在胃肠道中自发形成微乳,将多肽包裹在油滴中,既避免了酶解,又增加了与肠上皮的接触面积和溶解度。智能可吞咽设备是近年来最具颠覆性的技术方向,旨在绕过肠道屏障,直接将药物注入黏膜。RaniPill是一种“机器人胶囊”,口服后到达肠道,通过气球膨胀机制将载药微针快速注入肠壁,实现无痛给药。含奥曲肽的RaniPill在I期临床试验中表现出与皮下注射相当的生物利用度,且安全性良好,无严重不良事件。然而其挑战在于装置的复杂性和成本,以及患者对“体内微针注射”的接受度。4.药代动力学特征与负向选择框架
口服多肽药物的体内行为极其复杂,涉及从摄入到起效的多个动态过程。理解其药代动力学特征对于评估可开发性至关重要。口服多肽面临的首要挑战是极低的生物利用度。即便采用渗透促进剂,生物利用度通常仍仅为低个位数百分比,超过95%的给药剂量被“浪费”。这意味着只有高效能、低剂量的多肽才更适合口服给药,以限制原料药的浪费。考虑到多肽原料药的生产成本,这一因素在经济层面具有重要影响。药代动力学的第二个挑战是吸收的变异性。多肽在胃肠道的吸收往往是非线性的,受限于饱和转运机制或药物溶解动力学。食物的影响尤为显著,这种食物效应严重限制了用药便利性。同样,司美格鲁肽口服制剂也要求空腹服用,并需服药后至少等待30分钟再进食。第三个挑战是快速消除动力学。多肽的体内半衰期通常很短,从几分钟到几小时不等,这是其作为内源性信号分子的功能所需。进入体循环后,多肽易被肾脏快速清除或被肝脏代谢。这种快速的消除动力学要求药物具有极高的体内稳定性或特殊的组织靶向能力。针对这些挑战,近期研究提出了一个“负向选择”框架,用于早期识别不适合传统口服递送的多肽。三个反复出现的失败模式解释了历史记录:第一,暴露量不可行性:当低生物利用度与短消除半衰期相结合时,无论制剂技术多么复杂,都无法实现药物的有效积累;第二,变异性驱动的监管失败:当变异系数超过生物等效性要求所能接受阈值时,临床试验难以达到预期终点;第三,剂量攀升导致的胃肠道毒性和生产成本过高,在达到治疗之前就已失败。口服司美格鲁肽被分析为边界案例而非平台验证。其成功反映了一系列罕见属性的组合:超长半衰期、高效能、宽治疗窗口以及能够容纳低且可变吸收的时间整合型药效动力学特征。大多数多肽缺乏这一组合特征。口服奥曲肽在有限维持治疗适应症下的获批是另一个边界案例,同样揭示了这些约束条件。这一负向选择框架的核心结论是:口服给药的成败取决于分子药理学,而非制剂技术。口服递送应在药理学允许时采用,在药理学不允许时放弃——这是该领域可持续发展的先决条件。5.临床转化的经验教训与评估标准体系
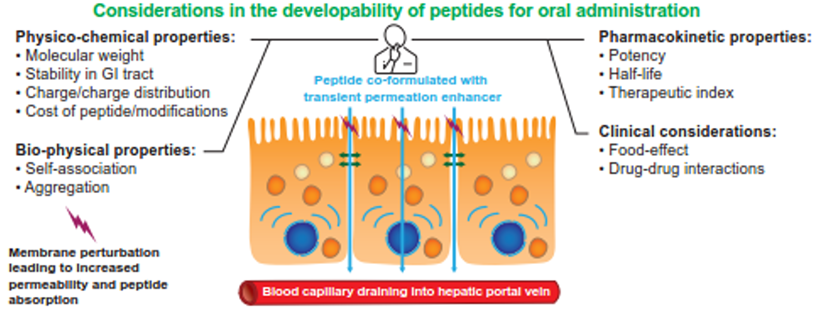
临床转化是检验药物开发策略的唯一标准。口服多肽领域充满了失败的教训和成功的曙光。口服司美格鲁肽的成功是里程碑式的事件。其成功归功于“分子修饰+制剂技术”的双重策略:分子层面经过修饰增加了稳定性;制剂层面利用SNAC技术显著提高了局部吸收。其生物利用度虽仍仅约0.5%-1%,但由于高效的药理活性和宽安全窗口,成功实现了临床转化。这一案例证明,低生物利用度药物只要具备足够的效价和安全性,仍有上市可能。口服奥曲肽采用Chiasma公司的TPE技术,通过在肠道特定部位短暂打开紧密连接促进吸收,于2020年获FDA批准,其血药浓度曲线与注射剂相当,证明了特定制剂技术在特定多肽上的可行性。环孢素A作为早期成功的环状肽案例,通过自乳化系统解决了溶解性和渗透性问题,其较高的亲脂性提示环状结构若能结合适当的亲脂性改造,可显著改善口服吸收。图4 口服多肽的可开发性考量然而失败案例同样值得深思。Oramed口服胰岛素尽管早期临床数据令人鼓舞,但在关键临床试验中未能达到主要终点,最终研发终止。失败原因可能包括胰岛素本身的稳定性与渗透性双重难题,制剂技术在复杂多变的人体胃肠道环境中无法保证稳定一致的吸收,以及糖尿病患者胃肠道排空速率差异大导致药效不可控。正如中学化学课上老师所传授的经验一样,结构决定性质,而化合物所呈现的性质不过是其结构的表象而已。这似乎有恰好验证了小学语文老师经常说的,良好的开端是成功的一半。口服多肽制剂能否达到一定的生物利用度和临床疗效由胃肠道固有的性质,制剂的设计以及多肽的特性所决定,而其中最关键可能就是咱们的化合物的性质了。以上种种似乎又都指向了化合物设计,强调了化合物设计的至关重要性。不知道以上推理是否正确,如果是正确且合理的,好像又恰恰说明了大道至简的这个道理!!!- Peptide-based therapeutics: quality specifications, regulatory considerations, and prospects
- Oral Peptide Delivery Reimagined: Molecular Barriers, Formulation Strategies, and the Rise of Computational Design
- Resmetirom and semaglutide in metabolic dysfunction-associated steatohepatitis (MASH): a comparative perspective
- Considerations in the developability of peptides for oral administration when formulated together with transient permeation enhancers
转载说明:本文系转载内容,版权归原作者及原出处所有。转载目的在于传递更多行业信息,文章观点仅代表原作者本人,与本平台立场无关。若涉及作品版权问题,请原作者或相关权利人及时与本平台联系,我们将在第一时间核实后移除相关内容。
 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库