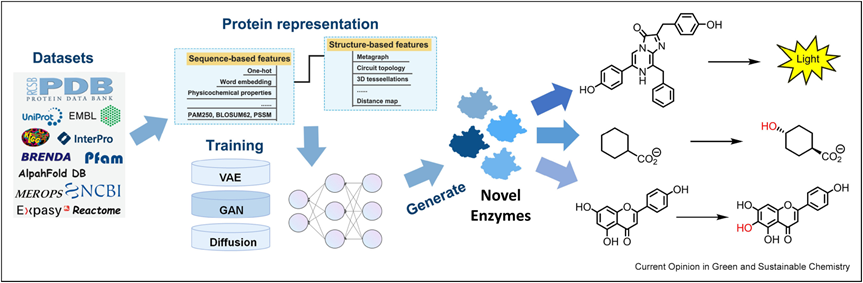
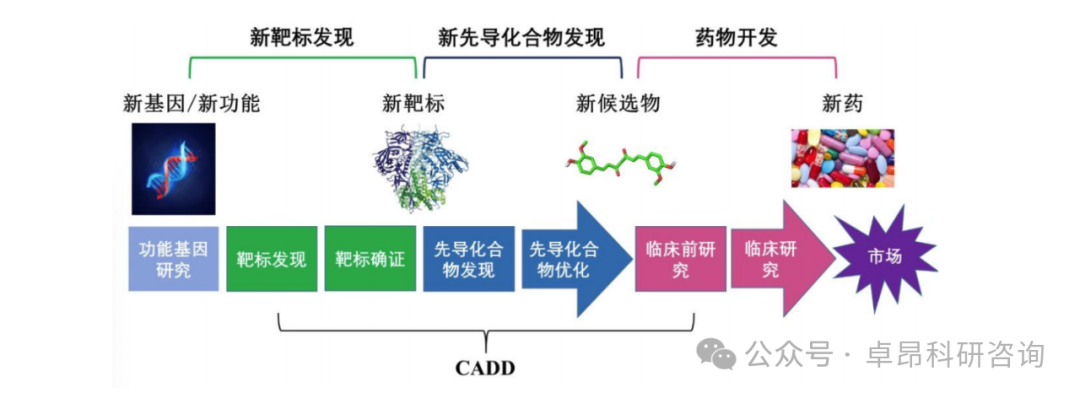
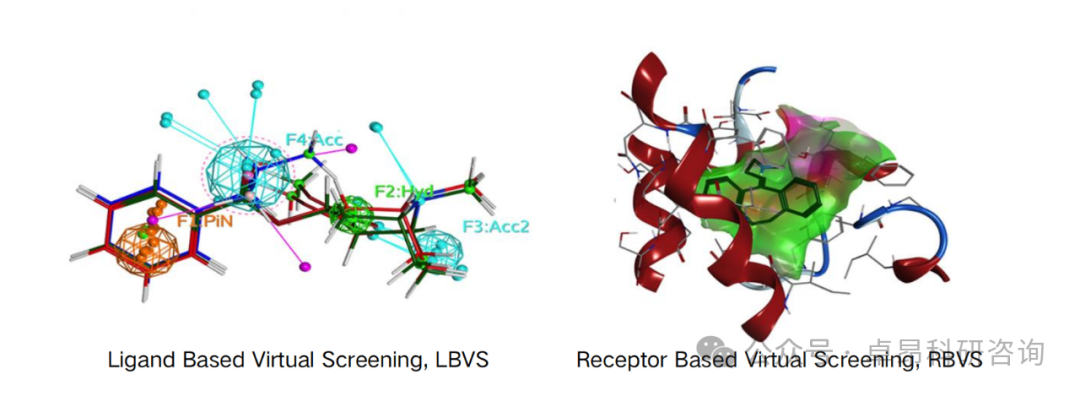
第一天:熟悉超算环境与蛋白质从头设计实践
1.环境搭建:Linux,VScode,Jupyter notebook*
a)超算的登录
b)Linux系统的常用shell命令:vim, ls, cd, less, rm等;
c)一些package安装的常用命令:pip, conda, source等。
d)Jupyter notebook的安装和使用。
e)VScode的基本配置:连接服务器;选择不同python版本的Interpreter;debug模式的使用等。
2.基础知识讲解
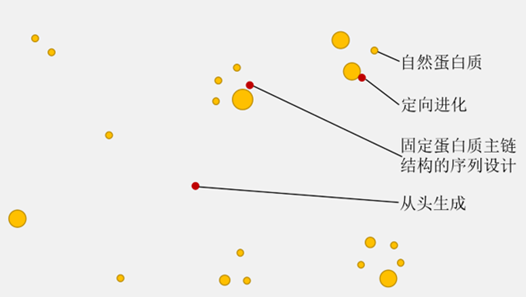
a)三类方法在不同程度上探索蛋白质序列空间:
i. 蛋白质定向进化(directed evolution)
ii. 固定蛋白质主链的序列设计(Fix-backbone protein design)
iii. 蛋白质的从头设计(De novo protein design)
![]()
b)关键数据库:RCSB PDB, SCOPe, CATH, UniRef, BFD等
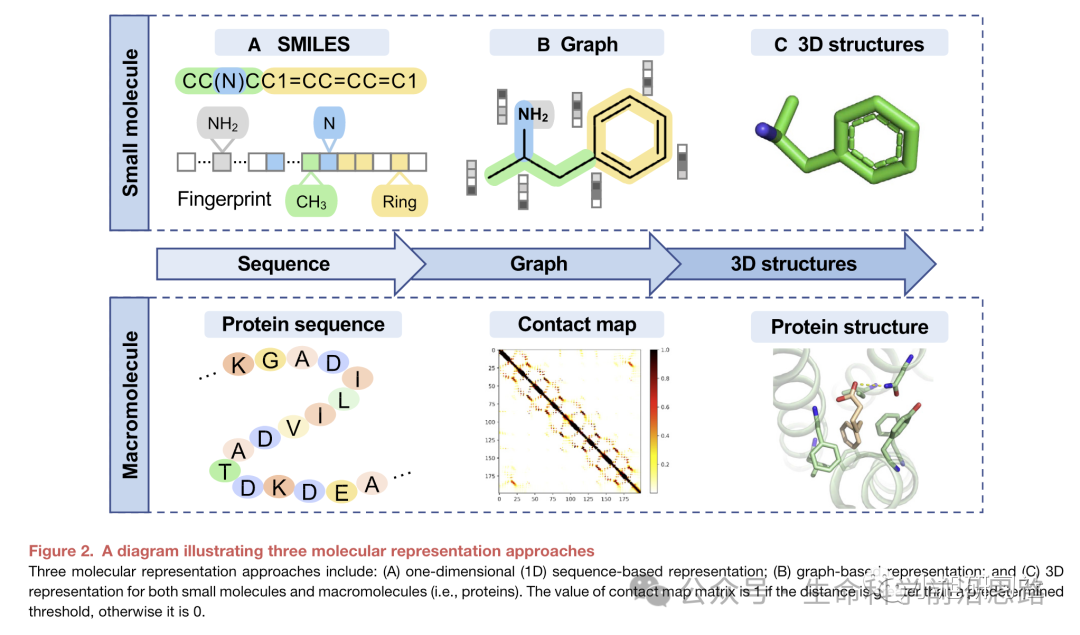
c)常见概念和名词: rotamer,scaffold, motif,domain,backbone,side-chain,apo和holo结构,
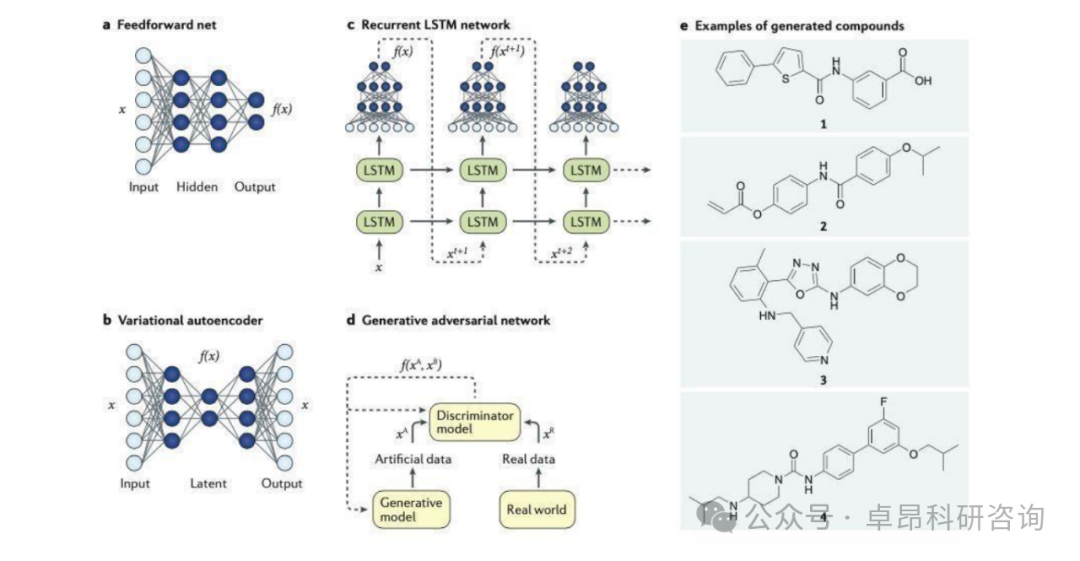
d)使用的不同模型的原理,transformer,diffusion模型,Flow Matching等。
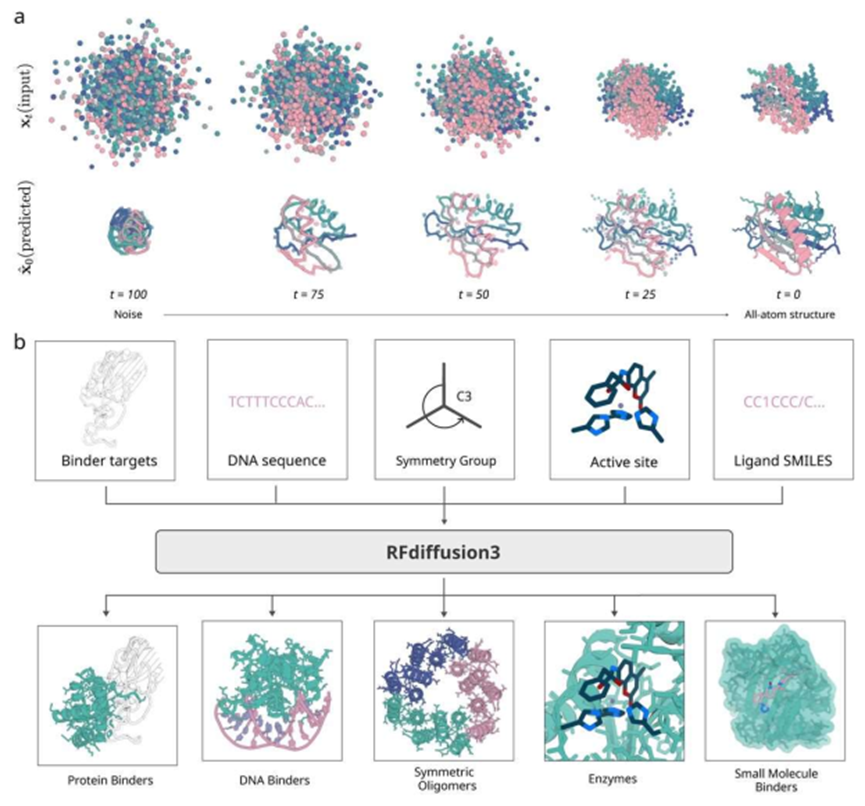
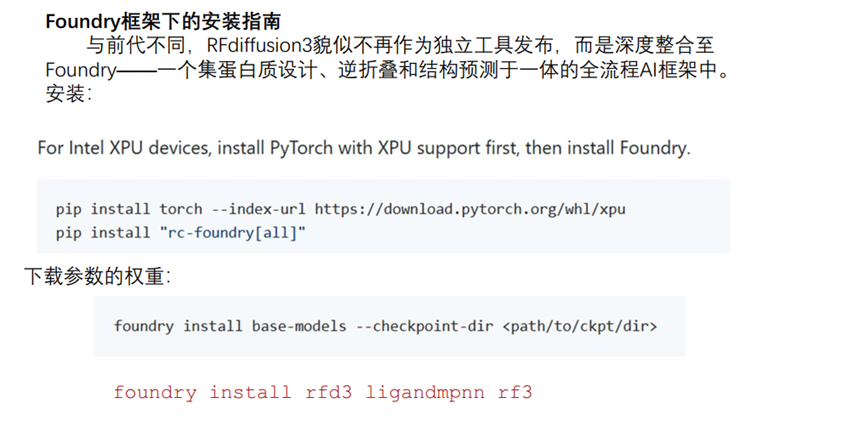
3.Rfdiffusion3+ProteinMPNN生成序列
a)Rfdiffusion3生成蛋白质骨架结构,ProteinMPNN精细的生成氨基酸序列。
b)Rfdiffusion3的安装实操
![]()
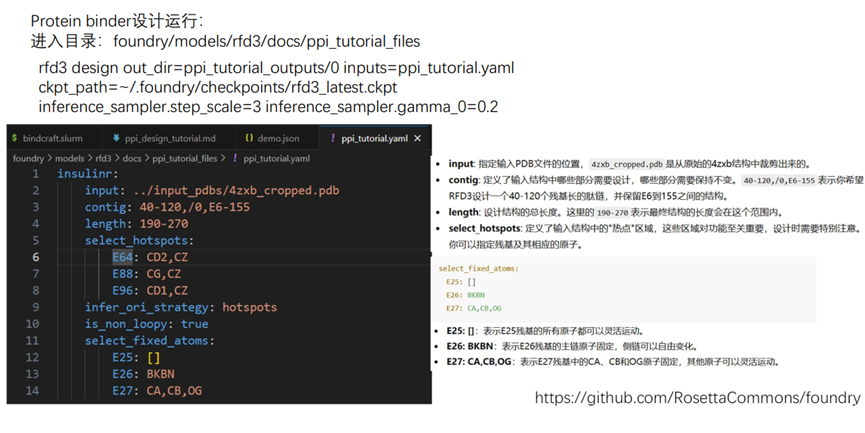
c)Rfdiffusion3的使用实操
![]()
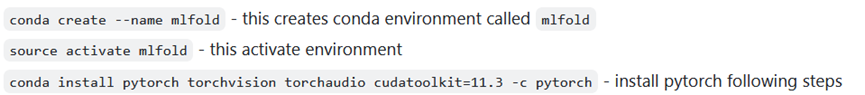
d)ProteinMPNN的安装实操
![]()
e)ProteinMPNN的使用实操

![]()
f)Rfdiffusion+ProteinMPNN生成序列,AphaFold2筛选序列。整体实操流程:
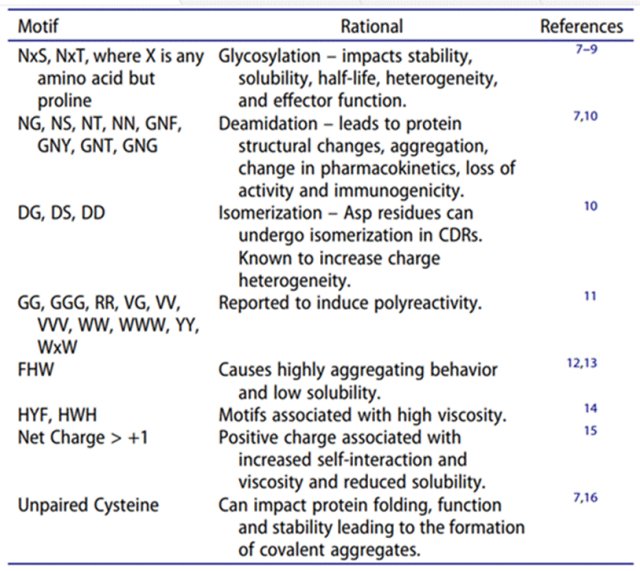
i. 计算SAP(Spatial Aggregation Propensity)的值,选择3-6个氨基酸作为hotspot,即结合位点;这里需要使用Rosetta进行计算,首先将安装rosetta,准备蛋白,再计算每一个氨基酸的SAP值,将SAP数值映射到结构上。选择hotspot位点。
ii. Rfdiffusion结构设计,生成~10000个蛋白质主链结构;
根据上面挑选得到的hotspot位点,更改相应的hotspot参数,生成新的结构
iii. ProteinMPNN-FastRelax进行序列设计,每一个主链结构两个对应的序列,共设计~20000个序列;
iv. 筛选:使用AlphaFold2预测设计结构,预测的置信度pAE<10,预测结构与设计结构的RMSD<1A,从中挑选95个进行实验验证。
4.其它的蛋白质设计方法的实操*
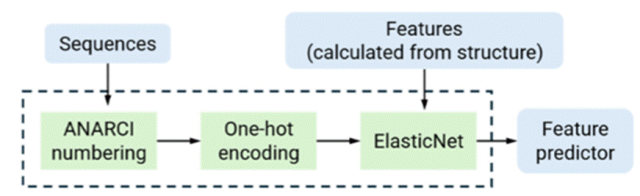
a)BindCraft——序列生成和筛选的自动化实现
BindCraft相比于Rfdiffusion+ProteinMPNN更加用户友好,一站式设计流程,序列的生成和筛选自动化实现。将讲解其中参数的设计和选择,如过滤序列条件、生成氨基酸的偏好性等。使用包括置信度评分(如AlphaFold2预测得到的pLDDT、ipTM)、物理指标(如Rosetta界面能量)和序列特征(如疏水性比例)进行筛选。
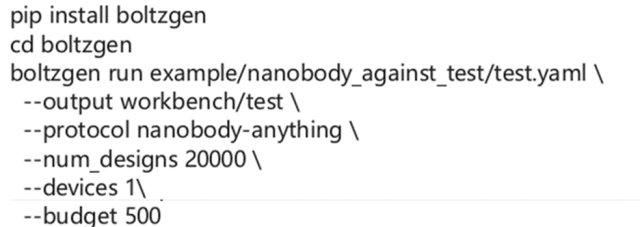
b)MIT开发的Bolzgen方法原理、安装使用讲解。
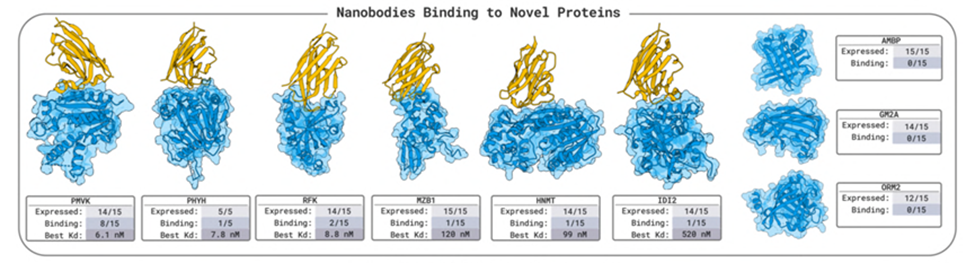
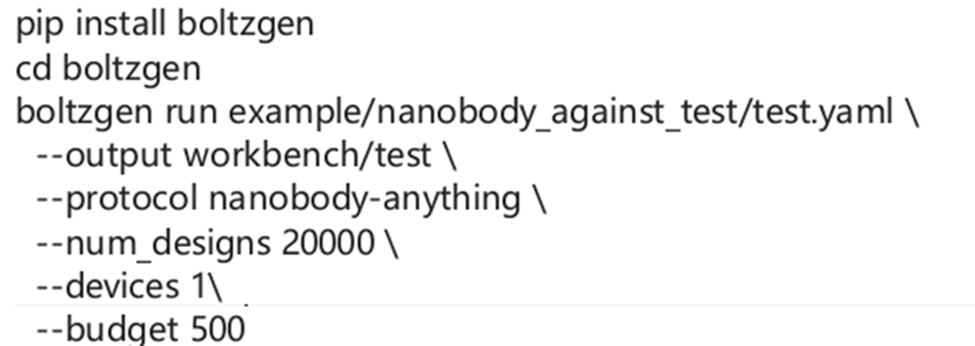
安装和使用boltzgen讲解,将详细讲解yaml配置文件的写法,以一个靶点为例,从头生成VHH与该靶点结合。
 c)PPIFlow:基于flow-matching的生成方法,原理,安装和使用方法。
c)PPIFlow:基于flow-matching的生成方法,原理,安装和使用方法。
![]()
第二天:蛋白质设计基础1——结构分析
![]()
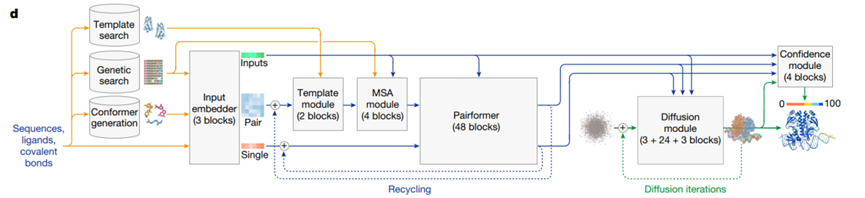
1.蛋白质结构预测方法
1)从CASP比赛结果来简述蛋白质结构预测方法的发展。基于能量函数 -> 接触图的应用 -> 端到端的预测结构(AlphaFold2)。
2)AlphaFold2的模型相比于以前的方法有什么改进
a)将基于MSA和基于模板的方法整合,使用注意力机制进行MSA信息和模板信息的相互交流。
b)以前提取MSA信息为计算协方差矩阵,AlphaFold2创造性的直接将MSA信息作为输入,将图像识别的算法转变成了自然语言处理算法,减少了中间处理过程中的信息损失。
3)AlphaFold3相比于AlphaFold2改进了什么,还有什么不足。
a)扩展到了多种生物分子的复合物结构预测,包括蛋白质-DNA、蛋白质-RNA、蛋白质-小分子,并使用扩散模型。
b)复合物组装与动态预测缺陷,抗体-抗原复合物结构准确度有待提高。
4)运行网页server上的AlphaFold3预测结构,https://alphafoldserver.com*
5)如何使用AlphaFold3预测蛋白质的糖基化,不同糖基化的类型的输入方法。
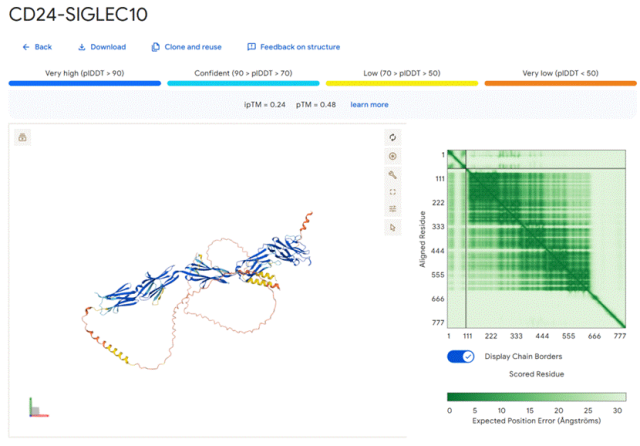
6)AlphaFold3输出结果分析,各项置信度指标的含义,以及如何判断预测的准确度,如pLDDT,ipTM,PTM,PAE。
7)本地部署和运行ColabFold,由于AlphaFold3在安装过程中需要下载大量资源,且不能商用,因此不演示AlphaFold3的安装过程,如有问题可以帮助解决。
a)git clone https://github.com/YoshitakaMo/localcolabfold.git
b)wget https://raw.githubusercontent.com/YoshitakaMo/localcolabfold/main/install_colabbatch_linux.sh
c)bash install_colabbatch_linux.sh
d)export PATH="/path/to/your/localcolabfold/colabfold
conda/bin:$PATH"
2.蛋白质结构分析和可视化
1)pdb文件的解读,每一行中的内容代表什么含义。
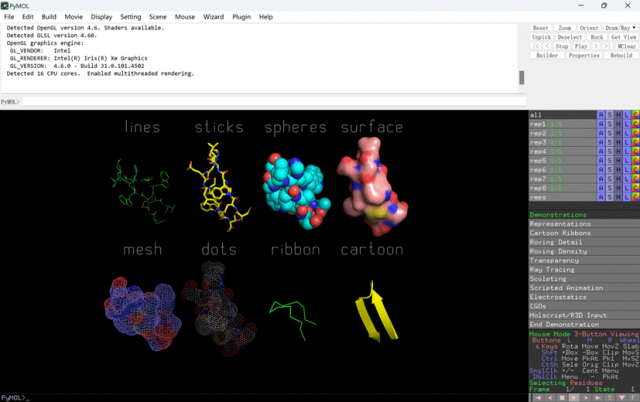
2)用 pymol 可视化蛋白质结构*
a)pymol的基础操作讲解
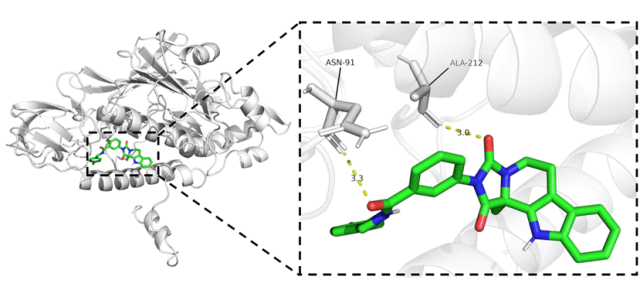
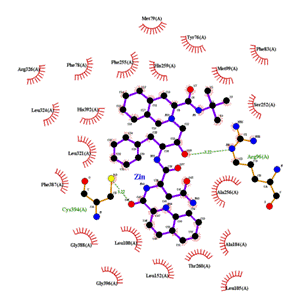
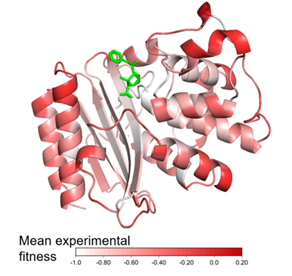
b)如何将实验值投影到结构图的颜色上,如何画出发表文章中好看的图
![]()
3)计算蛋白质结构中两个氨基酸的距离*
a)使用python的文本文件操作实现
b)使用python中biopython包实现
3.蛋白质结构相关物理性质的计算*
1)二级结构的分类和计算
2)溶剂可及表面积(SASA)的讲解及计算
第三天:蛋白质设计基础2——序列分析
讲解和实操:
1. 获得同源序列
1)了解不同蛋白质序列库,如UniRef90,UniClust30,Pfam等
2)了解不同工具原理并使用:NCBI BLAST,Jackhmmer,HHblits
3)给定一条蛋白质序列,比对序列库,生成多序列比对(MSA)*
从AlphaFold2的经典代码仓库中找到它的生成MSA的代码并学习(alphafold/alphafold/data/tools/jackhmmer.py)。
运行示例:jackhmmer --cpu 8 -N 2 -E 1e-7 query.fasta uniprot_sprot.fasta -o output.sto
2. 对MSA进行频率分析*
1)使用python的文本文件操作实现
2)使用python中biopython包实现
3)绘制序列Logo,可视化的展示每个位点的氨基酸频率和保守性
3. 序列的同源性计算和进化树的绘制*
1)不同同源性的计算方法及应用情景,氨基酸序列的identity和Similarity,BLOSUM62的介绍。
2)进化树的绘制
4. 基于序列相似性阈值划分训练集和测试集*
1)为什么要做?避免数据泄露
2)选择相似性度量方法
3)相似性矩阵的计算
4)划分数据集
5. 大规模蛋白质序列的聚类分析和去冗余*
1)为什么要做?防止过度学习某一类序列特征,消除序列偏差;也能防止训练过程中数据泄露。
2)聚类方法的选择,CD-HIT、MMseq2和Linclust
3)选择代表序列,去冗余
4)实际复现S2ALM这一模型文章中的聚类方法。mmseqs easy-cluster examples/DB.fasta clusterRes tmp --min-seq-id 0.7 -c 0.8 --cov-mode 1
第四天:蛋白质的大语言模型及其应用
 1.基础知识讲解
1.基础知识讲解
1)介绍蛋白质的语言模型(26字母语言模型->20氨基酸字母表,上下文依赖->氨基酸的共进化)
2)为什么要开发蛋白质大语言模型?1. 相比于结构或功能信息,序列信息更加海量;2. 蛋白质序列通过进化而来,可以学习蛋白质基本规律,折叠,共进化等
3)模型架构和基础理论:transformer,多头注意力机制,Bert,GPT,T5等
2.基于Bert架构的蛋白质语言模型
1)ESM系列(ESM-1b、ESM-1v、ESM2、ESM C)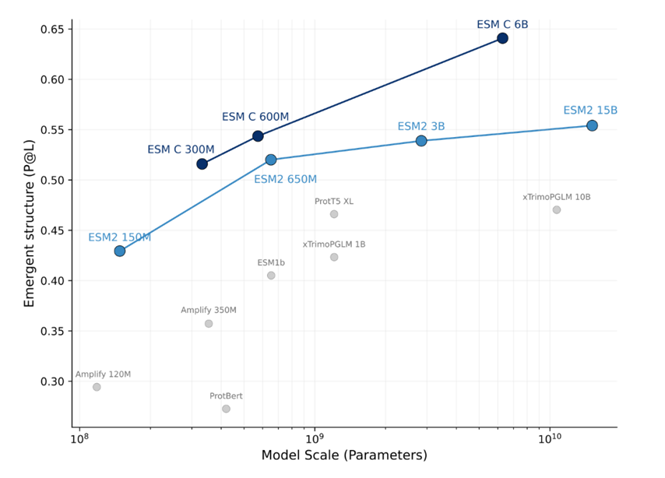 2)ESMFold:无需MSA信息的结构预测
2)ESMFold:无需MSA信息的结构预测
3)使用抗体序列库训练的语言模型:Ablang,AntiBERTy
3.类似GPT的生成模型ProGen1)36层Transformer解码器架构,包含12亿参数
2)引入“控制标签”(如蛋白质家族ID、功能属性)作为输入,生成蛋白质序列空间以外的新的蛋白质序列
3)成功生成新的溶菌酶
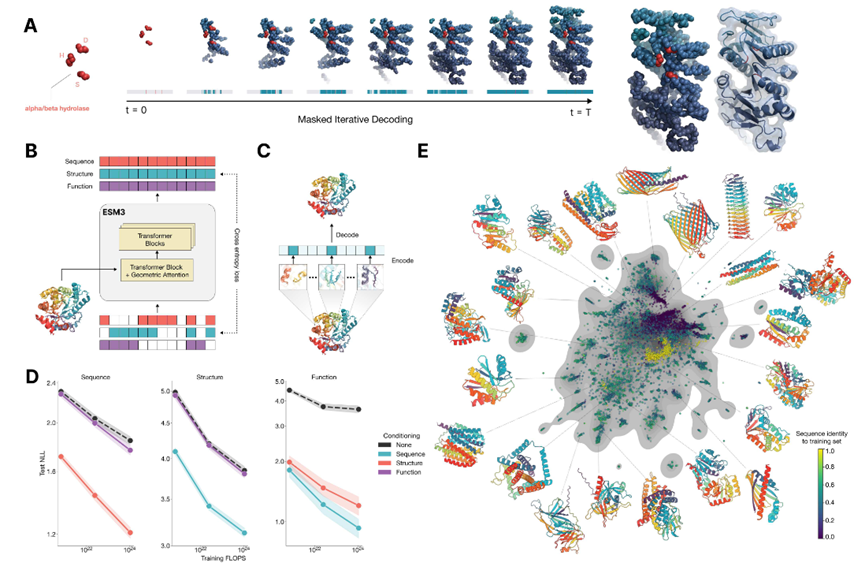
4.多模态的蛋白质语言模型ESM3
1)模型架构融合序列,结构和功能信息
2)相比于ESMFold,单体结构预测精度更好
3)基于多模态提示(序列、结构、功能关键词)设计新的蛋白质序列
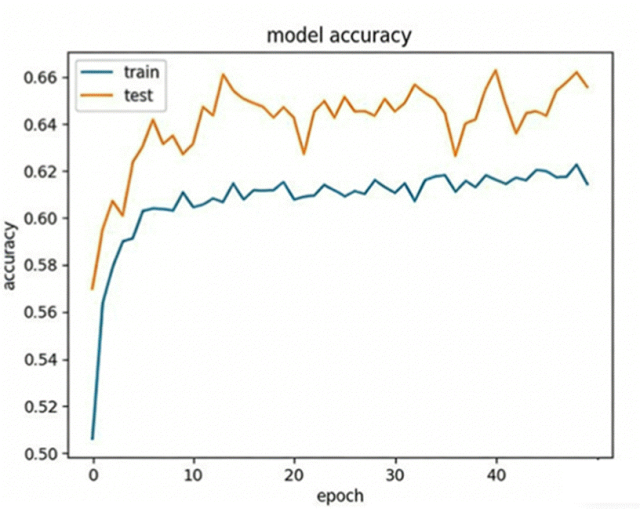
4)ESM3的安装,生成序列,快速结构预测。*5.蛋白质语言模型的应用和实战演练*
1)获得序列embedding以构建下游模型(Cell systmes文章举例),从文章github仓库中提炼序列embedding的代码并学习使用。https://github.com/fhalab/MLDE?tab=readme-ov-file#generating-encodings-with-generate_encoding.py,看懂代码中EncodingGenerator的类,将这个类方法用在我们自己的代码上,实现蛋白质序列的不同方式encoding,包括"onehot", "georgiev", “esm”系列模型。2)使用不同的蛋白质语言模型,零样本的预测蛋白质突变效应。3)给定少量的突变效应数据作为训练数据,训练模型,预测新的突变效应值。
第五天:深度学习辅助酶设计
1.基础知识讲解
酶的过渡态理论,theozyme,fitness landscape,epistasis
2.酶学性质预测
1.DLKcat与GotEnzyme数据库介绍
2.UniKP:利用预训练模型挖掘、改造Kcat
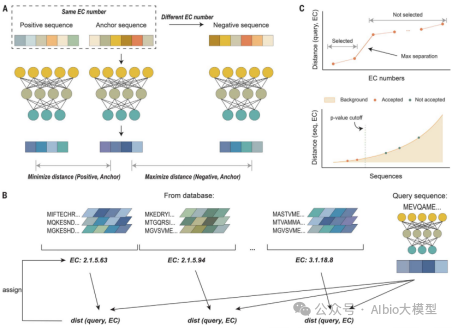
3.CLEAN:基于对比学习的EC号预测挖掘稀有脱卤酶
3.蛋白质热稳定性改造
1.MutCompute介绍
2.利用MutCompute改造PETase(Nature)
3.ThermoMPNN介绍与使用*
4.Pythia介绍与使用*
4.从Frances H. Arnold(2018年因在酶的定向进化领域的贡献获得诺贝尔化学奖)的工作看酶的定向进化方法的发展
1. 传统定向进化实验流程
2. MLDE(Mechine Learning Directed Evolution),学习序列与酶性能之间的映射关系,推荐新的突变组合(PNAS文章)
3.ftMLDE(focused training MLDE),主动学习流程,构建informative的训练数据(Cell Systems文章)。零样本突变效应预测挑选数据集,再通过小样本数据训练的策略微调。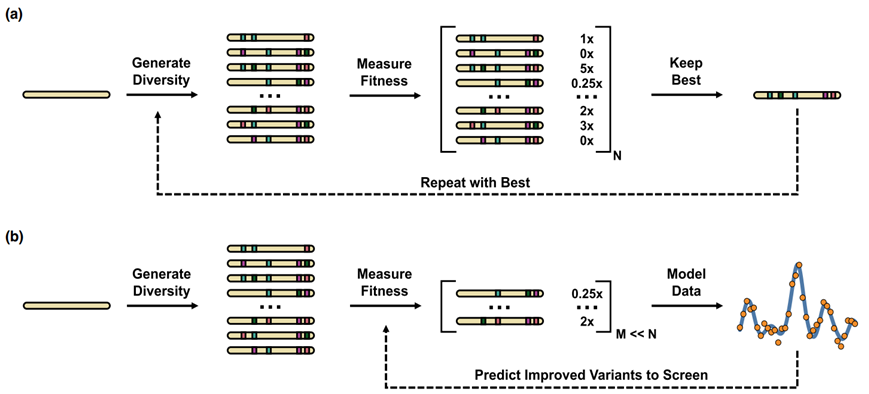 5.酶的从头设计
5.酶的从头设计
1.从头设计Diels-Alder催化酶
a)基于Rosetta的Inside-out策略(Science文章)
b)通过Foldit蛋白质折叠游戏改善结构问题(Nat. Biotechnol.文章);c)Foldit蛋白质折叠游戏的实践*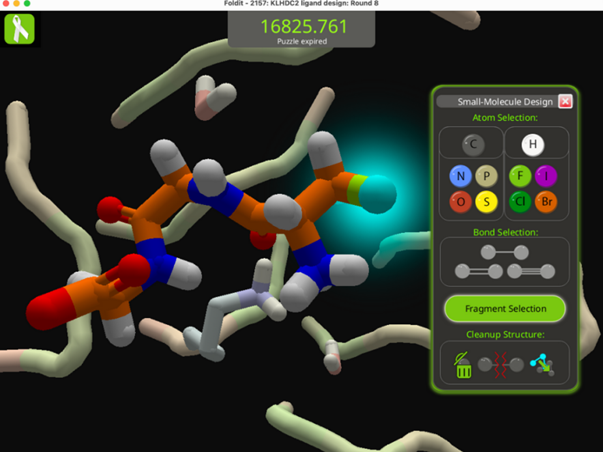 2.从头设计荧光素酶,Family-wide hallucination,基于该酶家族的结构幻化出新的结构(Nature文章)
2.从头设计荧光素酶,Family-wide hallucination,基于该酶家族的结构幻化出新的结构(Nature文章)
3.RFdiffusion+PLACER从头设计丝氨酸水解酶(Science文章)
6.利用预测结构的相似性,挖掘序列的新酶功能(复现顶刊cell文章)*
1. InterPro数据库中下载数据
2. TM-score计算结构距离
3. UPGMA结构聚类,画出进化树
4. 挑选序列
第六天: 蛋白质功能与互作预测;实验验证与AI模型训练预测闭环
1. 蛋白质功能预测:
1) 基础知识:
a) 基因本体论(Gene Ontology, GO),
b) MF/BP/CC,MF Molecular Function 分子功能;BP Biological Process 生物过程;CC Cellular Component 细胞组分。
c) GAF (GO Annotation File) 文件。
d) 本体文件来理解GO术语之间的层次关系。
e) 解析GAF,提取蛋白质ID和GO ID。
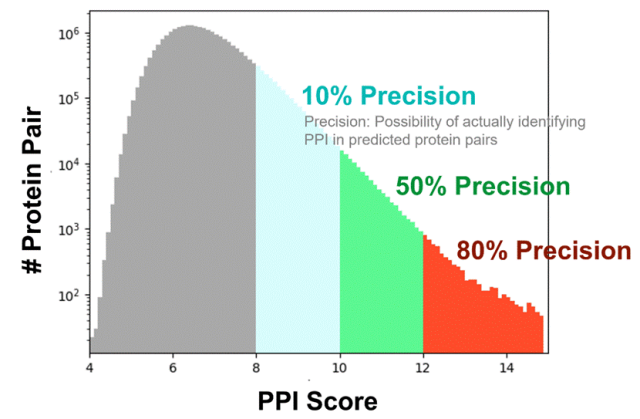
2) DeepGO-SE,通过蛋白质的语言模型提取序列嵌入,预测蛋白质的功能3) DPFunc:先用蛋白语言模型提取残基特征,再在接触图上用 GCN 学习结构信息,并引入结构域(domain)指导,最后把多层特征映射到 GO 图上,显著提升对罕见功能项和低序列相似蛋白的预测精度 4)Prot2Text-V2模型。Prot2Text-V2将图神经网络(Graph Neural Network, GNN)与大型语言模型(Large Language Model, LLM)融合到同一个编码器-解码器框架中,有效整合了包括蛋白质序列、结构和文本注释在内的多种数据,以自由文本形式输出蛋白质功能预测结果 5)ProteinKG65构建蛋白质知识图谱,基于Gene Ontology (GO) 和 UniProt 等权威知识库,将蛋白质的功能、结构、相互作用等知识组织成图谱形式,支持下游的机器学习任务,如蛋白质功能预测、表示学习、药物靶点发现等2.蛋白质相互作用预测:Science文章:使用更深的进化信号:omicMSA+新的深度学习网络:RF2‑PPI。在全人类蛋白质组中筛出一批高置信度的互作,用于补齐人类互作图谱、解释疾病突变和蛋白功能。
1.更深的进化信号:omicMSA
从约 30 PB 的未组装基因组/转录组数据里挖人类蛋白的同源序列,而不仅仅依赖 UniRef 等传统数据库。
构建omicMSA,使得每个蛋白的深度比常规模板 MSA 深 7 倍左右,协同进化信号显著增强。
2. 新的深度学习网络:RF2‑PPI
基于 RoseTTAFold2 框架开发了一个新的 PPI 预测网络 RF2‑PPI,用来快速估计两条蛋白是否互作以及界面大致形态。
为了训练 RF2‑PPI,构建了很大的数据集:从约 2 亿个预测蛋白结构中抽取各种结构域组合,构建了大规模的 DDI 训练样本,使训练集规模相比传统 PPI 结构数据扩大约 16 倍
筛选流程:
1. 人类蛋白集合
取约 19,500 个人类蛋白序列(UniProt 等),所有可能的配对约 2 亿对。文章中实际筛查约 2 亿对蛋白组合。
2. 构建深度 omicMSA
对每个蛋白,以及蛋白对,基于 30 PB 基因组/转录组数据构建 omicMSA,并对每个蛋白对生成配对 MSA(pMSA),用于协同进化分析和后续深度学习输入。
3. 快速预筛:共进化 / RF2‑PPI 粗打分
先用直接耦合分析(DCA)等共进化方法,结合 RF2‑PPI 对 2 亿对蛋白打一个“互作概率”分数(RFIntProb),过滤掉大部分不可能的组合。
他们在一个中间步骤里,从 4360 万对预筛后的蛋白对中,用RF2‑PPI 进一步筛选出约 190 万对 RFIntProb > 0.3 的候选。
4. 精细建模:AlphaFold2 复合物结构
对这约 190 万对蛋白,用 AlphaFold2(多聚体/复合物模式)进行结构预测,得到每一对的三维复合物模型以及一个基于界面质量的互作概率(AFIntProb)。
根据 AFIntProb 以及界面大小等指标选择高置信度互作。
5. 高置信度集的定义
在所有蛋白对中,他们最终在“完全无先验”的全 2 亿对筛选中得到 6,763 个高置信度 PPI;
进一步结合已有数据库(STRING、BioGRID、UniProt 里有物理互作证据的 115万对蛋白对),在有先验证据的集合上又识别出 21,960 个高置信度PPI。
综合各种来源和精度阈值,共预测出 17,849 个 PPI,预期精度约90%,其中 3,631 个此前实验未报道的新互作。
3. AI模型训练预测和实验闭环
以 EVOLVEpro 为例,实践计算–实验闭环:
● 选取少量已测序列(野生型 + 文献或少量自设计突变),测定活性。
● 用蛋白语言模型把序列编码成向量,训练一个初始的监督回归模型(序列向量 → 活性)。
● 设定允许的突变范围(允许 1–3 点突变、限定在特定位点/区域)。
● 在该空间内大规模生成候选序列(10^3–10^5),可结合 embedding 空间附近搜索、局部扰动等策略。
● 用回归模型对所有候选序列预测活性或综合评分。
● 依据主动学习策略挑出一小批要做实验的序列:
● 直接选预测值最高的 top‑k;或
● 结合预测不确定性、序列多样性等,使样本既“高潜力”又“信息量大”。
● 合成/构建这批候选序列,利用高通量实验(如流式、板读、NGS 条形码筛选等)测定真实活性。
● 得到新一轮“序列–活性”数据。
- ●将新数据并入训练集,重新训练或微调回归模型(PLM 一般保持不变)。●重复“生成候选 → 预测选样 → 实验验证 → 更新模型”的循环,通常 3–4 轮即可显著提升目标性能。
 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库