 五度妙笔
五度妙笔 企业透视镜
企业透视镜 API商城
API商城
 数据库
数据库周末文摘 | 基于ICH E6(R3)指导原则的制药企业临床试验数据治理实施策略

引用本文



颜崇超.基于ICH E6(R3)指导原则的制药企业临床试验数据治理实施策略[J].中国食品药品监管.2026.03(266):70-83.
基于ICH E6(R3)指导原则的制药企业临床试验数据治理实施策略
Implementation Strategy for Clinical Data Governance in Pharmaceutical Enterprises Based on ICH E6 (R3) Guideline
颜崇超
江苏恒瑞医药生物计量部临床数据科学中心
YAN Charles
Clinical Data Science Center, Biometrics Department, Jiangsu Hengrui Pharmaceuticals Co., Ltd.
摘 要Abstract
2025 年国际人用药品注册技术协调会(ICH)发布的《E6(R3):药物临床试验质量管理规范技术指导原则》终稿文件将“数据治理”独立成章,以“质量源于设计”和“基于风险的质量管理”理念重构全球临床试验数据管控逻辑。国家药品监督管理局已发布公告,明确自2026 年3 月31 日后实施的药物临床试验均适用ICH E6(R3)指导原则。面对国内制药企业在该指导原则落地过程中可能存在的核心理念悬空、业务适配不足、角色转型不清、新增环节缺乏标准等挑战,本文基于近百项国内外多中心临床试验的实践经验,系统解析了该指导原则的历史演进与全球革新逻辑,重点围绕盲态保持(含随机化)、数据全生命周期管理、计算机化系统验证三大核心模块,梳理了实操中的常见漏洞与风险防控重点。本研究构建了“文件、系统、流程、风险”四维落地路径,针对不同规模企业与项目类型设计了差异化推进策略,明确了存量项目与增量项目之间的合规衔接规则。同时,配套提出了稽查轨迹审核专项方案、关键角色调整要求及“可监控、可审核、可优化”的体系保障机制。本研究形成了从理念解析、痛点拆解、路径设计到保障机制的闭环逻辑,以期为制药企业提供兼具合规性与实操性的实施指引,助力中国创新药突破国际研发的合规壁垒,加速全球化布局。
In 2025, the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) released the final version of E6 (R3): Guideline for Good Clinical Practice, which establishes data governance as an independent chapter and restructures the global clinical trial data management framework based on the concepts of Quality by Design (QbD) and Risk-Based Quality Management (RBQM). The National Medical Products Administration (NMPA) has announced that the ICH E6 (R3) guideline will apply to all clinical trials initiated after March 31, 2026. Facing challenges in the implementation of this guideline among domestic pharmaceutical enterprises, including conceptual misalignment, insufficient business adaptation, unclear role transformation, and lack of standardized procedures for newly introduced processes, this paper draws on practical experience from nearly 100 domestic and international multicenter clinical trials. It systematically analyzes the historical evolution and global innovation logic of the guideline, and focuses on three key modules: blinding maintenance (including randomization), data life cycle management, and computerized system validation (CSV). Common operational gaps and key risk control priorities are also summarized. Furthermore, this study proposes a four-dimensional implementation framework encompassing documentation, systems, processes, and risk management. Differentiated implementation strategies are designed for enterprises of different scales and project types, and compliance transition rules between ongoing and newly initiated projects are clarified. It also provides a dedicated audit trail review (ATR) scheme, key role adjustment requirements, and a system assurance mechanism characterized as monitorable, auditable, and optimizable are proposed. By establishing a closed-loop framework from conceptual interpretation and problem identification to implementation pathway design and assurance mechanisms, this paper aims to provide pharmaceutical enterprises with practical and compliant guidance, thereby supporting Chinese innovative drugs in overcoming international regulatory barriers and accelerating global development.
关键词Key words
ICH E6(R3);数据治理;质量源于设计;盲态保持;数据全生命周期管理;计算机化系统验证
ICH E6 (R3); data governance; quality by design (QbD); blinding maintenance; data life cycle management;computerized system validation (CSV)

1 研究背景
国际人用药品注册技术协调会(The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use,ICH) 发布的《E6 :药物临床试验质量管理规范技术指导原则》(E6: Guideline for Good Clinical Practice) 历经3 次关键迭代,逐步构建了全球临床试验数据管理标准框架。1996 年ICH发布的E6(R1)确立了纸质化数据记录基础规则;2016 年更新的E6(R2)顺应电子化趋势,增补了电子数据采集(electronicdata capture,EDC)、电子签名等规范,但未突破“事后核查补正”的传统模式;2025 年定稿的E6(R3)实现了里程碑式突破,将“数据治理”独立成章,以“质量源于设计”(quality by design,QbD) 理念重构质量管控逻辑,以“基于风险的质量管理”(risk-based quality management,RBQM) 理念优化资源配置,精准回应了数字化临床试验普及、全球多中心协同深化及数据国际互认的行业需求[1-4]。
作为ICH 管理委员会成员,国家药品监督管理局积极推进E6(R3)指导原则的本土化实施。2024 年11 月,国家药品监督管理局食品药品审核查验中心公开征求E6(R3)指导原则附件2草案意见[5] ;2025 年3 月,国家药品监督管理局药品审评中心发布E6(R3)指导原则及附件1 的中文翻译稿并公开征求意见[6]。根据《中国新药注册临床试验进展年度报告(2024 年)》统计,2024年我国药物临床试验登记总量为4900 项,其中新药国际多中心临床试验337 项,占新药临床试验(2539 项)的13.3%[7]。然而,国内制药企业普遍存在计算机化系统使用率低、数据全生命周期电子化覆盖率不足、系统验证完整度有待提高等薄弱环节[8],这已成为部分创新药海外申报进程延误的重要原因。ICH E6(R3)指导原则数据治理框架的高质量落地,有望成为中国创新药突破国际合规壁垒的关键支撑[9-10]。
2 研究目的与范围
本研究基于笔者在制药企业临床试验数据治理领域的长期实践。笔者曾深度参与近百项覆盖肿瘤、高血压等主流治疗领域的国内外多中心临床试验,具体承担了数据管理、随机化设计与盲态保持工作,并完整参与了数据采集、清理、质量核查的全流程,以及临床试验项目管理系统(clinical trial management system,CTMS)、临床试验电子主文档(electronic trial master file,eTMF)管理系统、临床试验数据全流程管理系统(clinical data total management system,CDTMS)、EDC系统、随机化与试验药物供应管理(randomization and trial supply management,RTSM)系统及外部数据管理(external data management,EDM)系统等多类关键系统的开发、验证与运营优化。实践中,笔者通过推动电子化工具搭建数据全生命周期管理链路,将自动化流程嵌入EDC建库、数据清理、传输与电子签名等环节,探索智能化算法在风险预警、逻辑核查中的应用,逐步形成了“标准化筑基、自动化提效、合规化保障”的实操经验与量化管理体系,有效引领了行业发展[11-15]。
本研究基于上述实践经验积累,核心围绕临床试验全流程数据治理实务展开,结合国内制药企业在实施E6(R3)指导原则过程中面临的核心理念悬空、业务全链适配不足、角色转型不清、新增环节稽查轨迹审核(audittrail review,ATR) 缺乏标准、分层落地无路径等挑战,以“理论与实践深度融合、合规与实操无缝衔接”为核心方法,聚焦制药企业实际合规需求,旨在明确E6(R3)数据治理成功实施的关键要素,研究具体围绕以下3个方面。其一,解析数据治理战略重构与核心理念本土化落地逻辑。研究突破传统“数据管理单环节优化”的局限,从临床项目全流程视角(含方案设计、临床运营、数据管控、系统支撑)构建战略框架,将盲态保持、数据全生命周期管理、计算机化系统验证(computerized system validation,CSV) 及ATR 纳入战略范畴。结合企业实操痛点,如数据质量目标(data quality objectives,DQOs)前置缺失、风险分级模糊、ATR 空白等,拆解QbD 与RBQM 理念从“理论要求”转化为“内部流程”的关键路径,以解决“理念先进但实操脱节”的共性问题。其二,拆解全链路适配与核心要点专项落地方案。研究针对临床运营、数据管理、信息技术(information technology,IT)、质量保证、医学事务等多部门职能, 厘清E6(R3)新范式与传统模式的核心差异。聚焦盲态保持(含随机化)、数据全生命周期管理、CSV、ATR 四大核心要点, 对照E6(R3)数据治理相关条款逐条梳理实操漏洞,明确各环节操作边界、风险防控重点与合规判定标准,尤其针对ATR 这一新增环节,建立涵盖“内容、频率、责任、工具”的标准化体系。其三,构建分层落地与体系建设机制。研究结合企业规模(如大型或中小型企业)与项目类型(如国际多中心临床试验或国内临床试验项目),设计分阶段推进、可动态调整的差异化合规路径,界定存量项目补正规则与增量项目前置要求。同时,配套“可监控、可审核、可优化”的体系建设机制,通过量化关键绩效指标(key performance indicator,KPI)、常态化监控与持续改进,着力解决“条款难以转化、落地缺乏保障”的核心痛点。
本研究范围聚焦制药企业在ICH E6(R3)指导原则框架下的数据治理核心模块,边界明确:覆盖场景方面,涵盖肿瘤、高血压等主流治疗领域创新药的临床试验,兼顾传统纸质数据与电子化数据场景, 重点关联盲态保持、数据“产生- 采集- 审核-传输- 归档- 销毁”全链路,以及支撑该链路的各类计算机化系统(如EDC 系统、RTSM 系统、CTMS 等)的合规性。排除范畴方面,本研究不涉及伦理审查、知情同意、受试者招募等非数据治理领域内容,仅围绕数据治理单一领域深耕,以确保问题分析与方案设计精准贴合相关企业在理念落地、流程优化、系统验证、ATR 等方面的实际需求,从而为后续实践提供针对性指引。
3 ICH E6 系列版本指导原则数据治理的历史演进与全球革新
3.1 历史演进:从“附属环节”到“核心支柱”
ICH E6 系列版本指导原则的迭代本质上是数据治理定位与责任体系的双重升级,其核心差异体现在管理逻辑、责任界定与行业适配性的深度变革(表1)。

E6(R3)的突破性在于:一是将数据治理从“被动响应型核查”升级为“主动设计型管控”;二是将责任体系从“分散式提及”迭代为“全周期权责矩阵”。二者共同支撑数据治理成为临床试验合规核心支柱[2-4]。
3.2 全球革新:核心理念、核心要点与责任体系的三维突破
相较于ICH E6(R1)及E6(R2)聚焦于确立“基础合规底线”,E6(R3)立足全球临床试验数据治理新需求,从“理念底层重构、要点实操细化、责任闭环落地”3 个维度实现系统性革新,形成“主动防控、可落地、可追溯”的完整数据治理体系。这三大维度呈现出“理念锚定方向、要点拆解路径、责任保障落地”的层层递进关系[16]。其中,理念革新深度承接了ICH《E8(R1):临床研究的一般考虑》及《E9(R1):临床试验中的估计目标与敏感性分析》的核心逻辑,实现了从“被动合规”到“主动控质”的范式转变[17-19]。
3.2.1 理念革新:“质量源于设计”与“基于风险的质量管理”
ICH E6(R3) 通过QbD、RBQM 与DQOs 的协同实施,构建了“理念、量化、执行”的闭环逻辑。QbD :从“事后纠错”转向“源头控质”,在方案设计阶段即明确“主要疗效终点数据必须100% 溯源至原始病历”等要求,并同步嵌入EDC 系统逻辑核查规则。RBQM :按数据对试验参与者安全、试验结论可靠性的影响程度分级管控,将资源向关键数据与流程倾斜(表2)。DQOs : 作为串联QbD 与RBQM 的量化载体,在方案设计阶段同步设定完整性、及时性、准确性等指标。例如,某糖尿病Ⅲ期临床试验设定“糖化血红蛋白数据录入误差率≤ 0.1%”的目标,配套系统自动校验结合100% 原始数据核对,使该数据错误率从5.3% 降至0.8%。协同逻辑:QbD 定方向、DQOs做量化、RBQM 配资源,形成全流程主动质量控制闭环。

3.2.2 核心要点细化:制定全场景可落地的操作标准
ICH E6(R3) 针对数据治理核心场景,以全流程风险管控为核心,细化了操作标准、责任界定、记录要求及与前版差异,确保每个要点均与理念革新深度绑定。
盲态保持:从“单一保护”到“随机化- 盲态全链路联动管控”。相较于E6(R2)的原则性提及,E6(R3) 明确要求将盲态管控嵌入系统设计、权限分配、流程执行、风险处置的全流程环节,形成“技术屏蔽、责任隔离、风险可控”的闭环体系。系统设计层面,EDC 系统需与RTSM系统的盲态管理深度联动,自动屏蔽敏感信息,严格分配用户权限。责任与风险管控层面,需在临床试验启动阶段明确设盲计划、揭盲人员与流程,对非盲人员实施物理隔离、签署保密承诺及全程审核,并及时归档。
数据全生命周期管理:从“环节碎片化”到“8 环节可追溯性、清晰性、及时性、原始性和准确性(ALCOA+)闭环”。E6(R3)首次将数据的“ 生成- 采集-审核- 更正- 传输- 确认- 保留- 销毁”全链条8 个环节纳入ALCOA+ 标准框架。在采集环节,需同步绑定核心元数据,以确保源头可追溯。在传输环节,跨系统数据流转必须执行一致性校验流程,如肿瘤影像学数据传输前生成唯一的消息摘要算法第5 版(Message-Digest Algorithm 5,MD5)校验码。在其他环节,审核与更正需留存操作轨迹与审批记录,确认环节需双人交叉验证,保留环节需存储于安全合规的服务器,销毁环节需执行“审批-擦除- 回执”流程。
CSV :从“基础功能测试”到“ 全生命周期V 模型验证”。相较于E6(R2)仅聚焦“功能可用”,E6(R3)要求采用《良好自动化生产实践指南第5 版》(Good Automated Manufacturing Practice 5,GAMP5) 框架下的V 模型实施系统全生命周期验证,覆盖“需求定义、设计、开发、测试、部署、维护、下线”全链路,即在需求定义阶段,需制定可验证、可追溯的用户需求说明(user requirements specification,URS)。全链路验证需遵循“左链开发、右链测试”的V 模型逻辑[13],涵盖系统级验证与项目级测试(图1)。CSV 需符合美国《联邦法规汇编》(Code of Federal Regulation,CFR) 第21 篇第11部分(21 CFR Part 11)、《欧盟药品法规汇编》第4 卷《附件11 :计算机化系统》(Eudra Lex Volume 4 Good Manufacturing Practice Annex 11: Computerised Systems)等国际公认的监管标准[20-22]。在文档闭环阶段,需形成“纵向贯通、横向关联”的文档体系,所有文档需留存至系统退役后5 年。
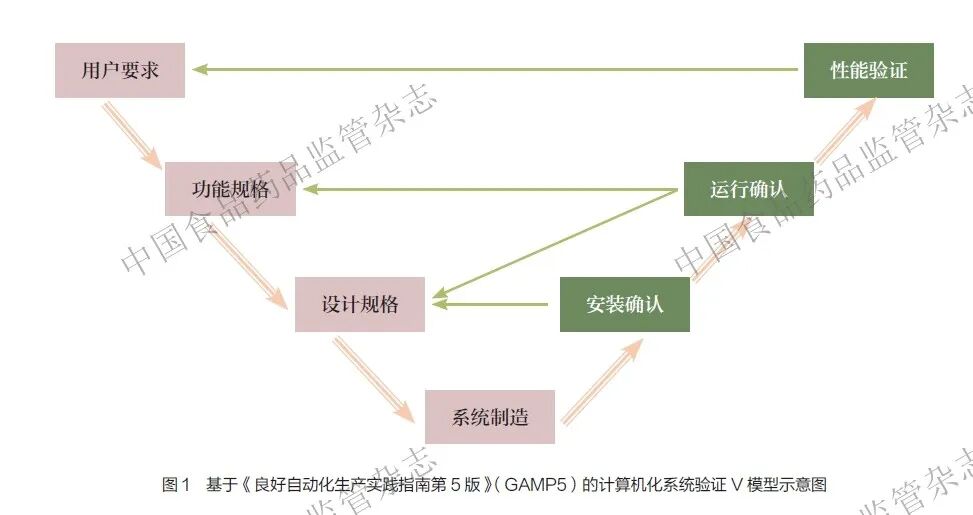
3.2.3 责任体系:构建申办者主导、研究者守源、多方协同的责任闭环
ICH E6(R3) 明确了数据治理的核心责任主体为申办者与研究者,其中申办者的“最终责任人”角色被重点强化,同时要求其与研究者协同,实现从源头把控、过程管控到结果闭环的责任协同。
申办者:作为数据治理的“总设计师”与最终责任人。E6(R3)明确申办者对临床试验数据的质量和可靠性承担“最终且不可转移的责任”。无论数据治理工作是自主执行还是外包,申办者均需通过“内部体系构建”与“外包全流程管控”实现责任落地。自主执行场景下,需建立流程标准化、质控节点化、评估量化的内部管理体系。外包管控场景下,需通过准入审核、过程监控、结果审核、违约处置的全流程管控,保留最终决策权。同时,申办者负有对研究者“赋能支持+ 评估监督”的责任。
研究者:作为数据质量的“源头守护者”与一线执行者。E6(R3)将研究者定位为“原始数据合规性第一责任人”,其核心职责聚焦于原始数据记录、电子病例报告表(electronic case report form,eCRF) 填写、盲态保持三大环节。对于原始数据,需确保其记录及时、准确、可追溯,修改需遵循相关规范。对于eCRF,需及时完成录入,核对数据准确性,及时回应数据质疑。在盲态保持方面,需严格执行揭盲审批流程,严禁泄露盲态信息,同时培训团队成员掌握相关规范与要求。
4 ICH E6(R3) 指导原则数据治理实施过程中的核心挑战
4.1 理念落地挑战:“质量源于设计”与“基于风险的质量管理”的实操挑战
4.1.1 “质量源于设计”落地不足:数据质量目标前置缺失
DQOs 需在方案设计阶段预先明确。然而,目前多数企业实践中仍存在“先采集数据,后补设DQOs”的现象,并常以“数据质量达标”等模糊表述替代量化指标,导致数据管理“无标可依”[23-24]。在数字化临床试验场景下,部分企业缺乏远程设备数据质量标准,后期常需多轮校准,甚至可能影响疗效与安全性结论的可靠性[25]。
4.1.2 “基于风险的质量管理”应用模糊:分级标准失配与资源错配
RBQM 的核心在于“ 风险分级与资源精准匹配”[26]。但实践表明,许多制药企业尚未建立适配数据治理场景的风险分级标准。例如,“实验室数据正常值范围”作为判断指标临床意义、界定不良事件的关键依据,理应被列为高风险管控项,但行业内普遍存在正常值范围提供滞后与风险应对策略缺失的问题[27]。某制药企业开展的一项肿瘤多中心临床试验案例颇具代表性:该项目覆盖国内12 家临床研究中心,1家中心实验室的数据被录入EDC系统后,仍需等待2~4 周才能获得其对应的正常值范围;而数据管理未将其列为高风险管控项,因而未及时调配资源跟进。待正常值范围补全后发现,约10%的丙氨酸氨基转移酶(alanineaminotransferase,ALT)数据未被及时清理,需重新核查修正,造成了不必要的成本浪费与项目时间延误。这种风险分级标准失配与资源错配并存的问题,本质是相关企业未将数据对临床试验结论的影响程度纳入风险分级核心维度,而仅依赖数据类型进行简单划分,违背了精准管控的核心逻辑。
4.2 业务适配挑战:三大核心模块的执行挑战
数据治理核心场景存在诸多实操挑战,本研究对其核心问题、产生原因以及防控措施进行了归纳(表3)。

5 ICH E6(R3) 指导原则数据治理三大核心模块的落地路径
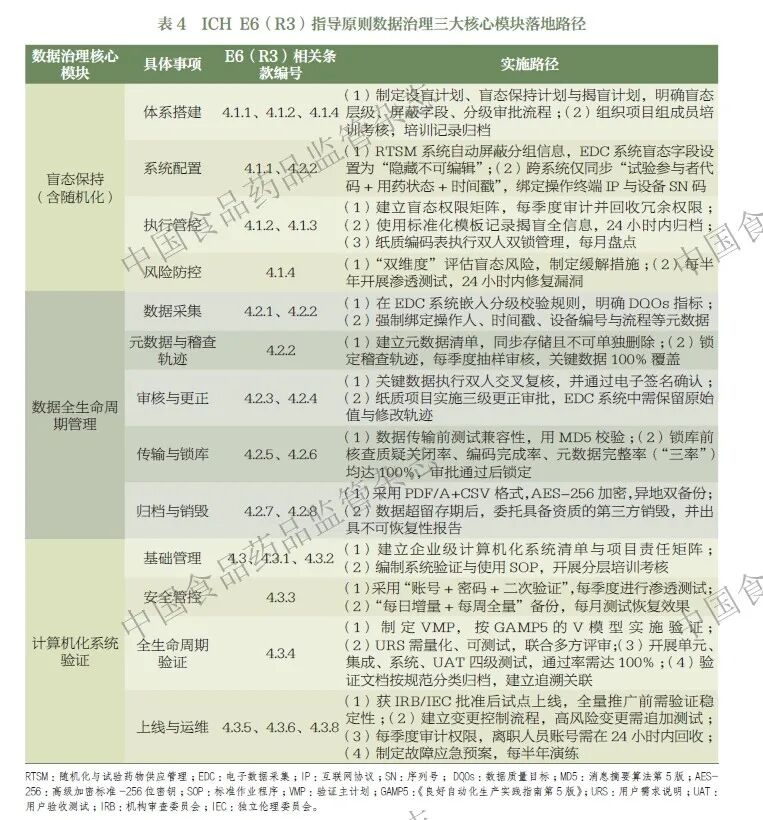
针对临床试验中普遍存在的盲态保持不规范、数据全生命周期管理存在短板、CSV 不完整3 类问题,本研究结合E6(R3)条款要求,从文件、系统、流程、风险4 个维度, 为三大核心模块构建了可直接落地的实施路径(表4)。
6 制药企业实施ICHE6(R3) 指导原则数据治理的方案
ICH E6(R3) 指导原则中的数据治理以“QbD+RBQM”为核心,实现了对传统临床试验数据管理模式的范式重构[28-32]。其落地不仅要求企业在人员能力、系统功能、流程逻辑上完成适配升级,更需结合自身规模与项目类型制定差异化推进策略,并通过体系化机制确保合规成效不断优化。本研究围绕战略顶层设计、核心差异对比、关键角色调整、ATR 审核专项、分层推进策略、体系建设机制6 个维度,系统梳理落地实施的影响与完整方案,形成“影响分析- 执行路径- 保障机制”的闭环逻辑。
6.1 战略顶层设计:从“单一环节合规”到“全流程主动控质”
ICH E6(R3)指导原则中的数据治理, 并非仅针对数据管理单一环节的优化, 而是对临床项目全链条(含项目设计、临床运营、盲态保持、数据全生命周期管理、CSV)的战略重塑。企业需突破传统“部门分割、环节脱节”的局限,从临床项目全局视角, 明确E6(R3) 模式与传统模式在核心维度上的本质差异,同步将DQOs、盲态保持、数据全生命周期管理、CSV 这4 类关键要求嵌入战略框架, 为全流程转型奠定基础[33-34]。本研究对具体战略维度的对比与落地核心要求进行了归纳(表5)。

6.2 核心差异对比:明确E6(R3)模式与传统模式的核心适配方向
ICH E6(R3) 指导原则的数据治理以“全流程主动控质”为核心,覆盖临床试验全流程(方案设计、执行、数据管控、系统支撑等)及相关部门(临床运营、数据管理、盲态保持、IT、质量、医学等),将盲态保持、数据全生命周期管理、CSV 三大核心模块融入各环节,与传统模式形成了系统性差异(表6)。

6.3 关键角色调整:适配ICH E6(R3)的职责升级
ICH E6(R3) 指导原则要求各关键角色突破传统工作边界,在职责、技能、工具、文档上完成系统性调整,且需适配不同规模企业的资源与能力。本研究对各关键角色的具体职责新增/ 强化内容、适配策略及核心产出文件进行了归纳(表7)。

6.4 稽查轨迹审核专项:新增环节标准化落地
ATR 是ICH E6(R3) 指导原则新增的关键管控节点,通过对稽查轨迹的深度审核,彻底改变传统实践中“仅留存不审核”的模式。相关企业需从内容、频率、工具、责任等方面建立标准化体系。本研究对具体落地要求进行了归纳(表8)。

6.5 分层推进:适配不同规模企业的差异化策略
6.5.1 大型制药企业:“专项团队+ 自主管控”模式
大型制药企业可组建跨职能专项团队,核心成员应具备国际合规知识与风险管控复合能力。对于增量项目, 全流程需贴合ICH E6(R3)指导原则要求;对于存量项目, 需补充ATR、元数据归档等合规措施。相关企业可选择代表性项目进行试点,形成内部实操指南,以实现标准复用。
6.5.2 中小型制药企业:“轻量化工具+ 外部协作”模式
中小型制药企业可优先选用自带ICH E6(R3) 指导原则合规功能的EDC 系统、RTSM系统等工具;联合合规咨询机构、合同研究组织(contract research organization,CRO)制定整改计划,重点突破核心环节;可优先保障国际多中心申报项目合规性,对于国内非关键项目,可分阶段落地,聚焦核心数据与流程。
6.5.3 存量项目与增量项目衔接原则
对于存量项目,可沿用原有流程,补充ICH E6(R3)指导原则合规报告,说明ATR、元数据管理补正措施。对于增量项目,可在启动前完成“三前置”,即DQOs 确认、盲态管理计划批准、CSV 报告备案,未达标不得启动。
6.6 体系建设:构建长效保障机制
6.6.1 设定关键绩效指标
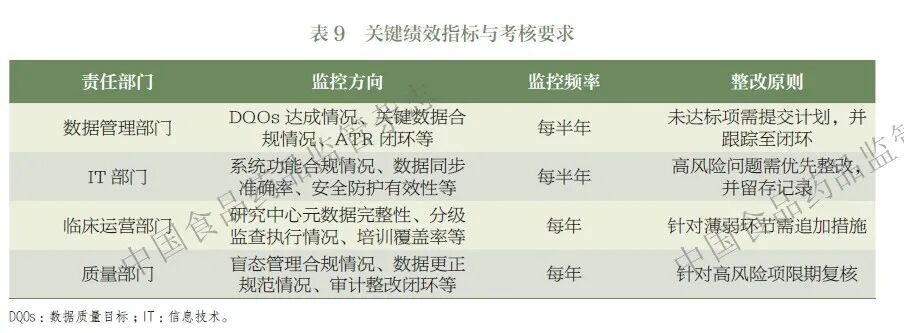
制药企业可围绕ICH E6(R3)指导原则的关键要求设定量化指标(表9),并纳入相关部门考核,从而推动从“无标准管控”到“可衡量合规”的转变。
6.6.2 开展常态化内部审计
建议制药企业以风险导向为核心开展常态化内部审计,推动从“被动迎检”到“主动自查”的转变。审计范围方面,可聚焦ICH E6(R3)指导原则的核心环节,包括DQOs 执行、盲态管控、ATR 记录、系统验证、元数据归档,覆盖关键项目与核心数据。审计原则方面,可按项目类型分层抽样,对关键数据(如严重不良事件、疗效终点)全量核查,确保重点突出、风险可控。整改要求方面,可出具审计报告并标注风险等级,对高风险项需限期整改并复核,避免传统模式下“审计无结果、问题反复出现”的弊端。
6.6.3 推动持续优化
制药企业可建立定期复盘机制,实现从“静态合规”到“动态适配”的转变。在复盘重点时,可汇总监控指标未达标项、审计发现的问题、实操中的痛点,梳理在理念落地、角色执行、流程衔接等方面存在的短板。在优化方向上,可及时更新内部文件(如实操指南、风险矩阵),调整管控措施;迭代培训内容,聚焦新出现的合规难点,确保团队能力与ICH E6(R3)指导原则要求同步升级,避免“一次性合规后体系僵化”的风险。
7 小结
ICH E6(R3) 指导原则数据治理的实施,本质上是推动临床试验质量管理从“被动合规补正”向“主动设计控质”的范式转型,其核心以QbD 与RBQM为双引擎,实现“合规、提质、降本”三重价值目标。本研究明确了该指导原则成功实施的关键逻辑:战略层面,完成全流程主动控质体系重构;业务层面,突破部门壁垒,实现全链条职能协同;落地层面,立足资源禀赋,实施分层策略。同时, 以量化KPI、开展常态化内部审计、持续优化机制,共同保障体系运行的长效性。
本研究构建的落地路径与体系机制,旨在针对性解决制药企业理念转化、流程适配、角色调整等实操难题,力求贴合我国医药行业的监管政策导向与创新药国际化发展需求。未来, 随着ICH E6(R3)指导原则本土化实施的深入推进,本研究形成的框架可为监管部门提供行业实践参考,为制药企业搭建合规管理框架,从而推动我国临床试验数据质量与国际先进标准接轨,为医药产业高质量发展与全球化竞争提供支撑。
致谢
本研究得到江苏恒瑞医药股份有限公司的平台与资源支持,感谢江苏恒瑞医药股份有限公司临床质量保证部负责人胡娟女士的专业帮助,感谢江苏恒瑞医药股份有限公司临床数据科学中心陈昕、别林、彭锐、周磊、颜怀海、魏鑫、周尔康与黄小莉等同事的协作支持,同时也感谢行业同仁分享的实践经验。
作者简介
颜崇超,博士,江苏恒瑞医药生物计量部临床数据科学中心,副总经理。专业方向:临床试验数据治理、临床数据管理、临床试验随机化与药品管理、临床试验一体化平台与计算机化系统验证
参考文献:略




编辑:李丹
审核:赵燕宜






