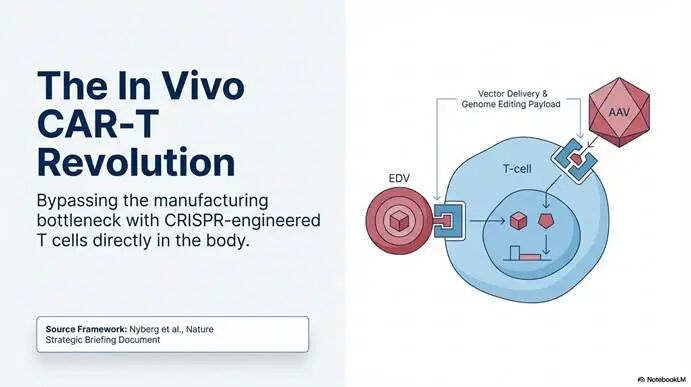
走向“即用型”细胞治疗:体内CAR-T递送系统的开发现状与未来挑战
发布时间:2026-04-20来源:药事纵横
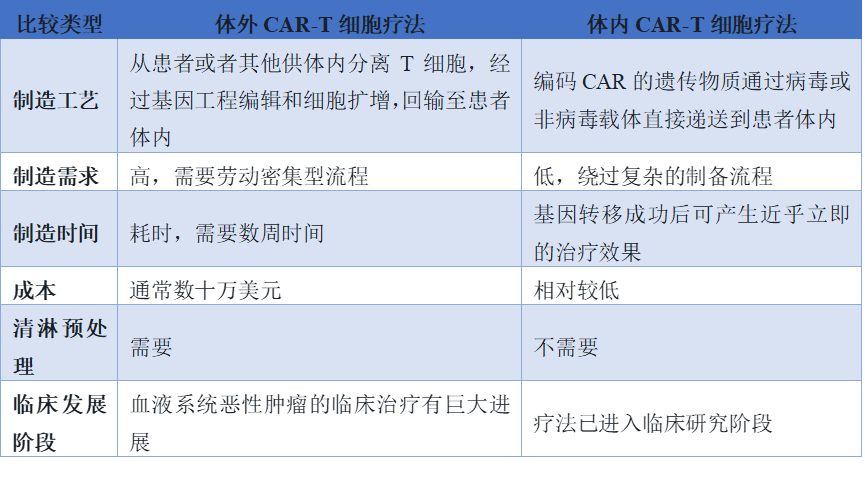
肿瘤的传统治疗手段主要包括手术、放疗和化疗。尽管这些常规方法在过去的几十年中取得了长足进展,但对于晚期肿瘤患者而言,由于肿瘤的高度异质性以及全身毒性的限制,传统疗法的治疗效果往往遭遇瓶颈。与传统治疗思路截然不同的免疫治疗异军突起,凭借其独特的作用机制,相关药物获批数量持续增长,多款免疫治疗策略正处于临床前研发或临床试验阶段。在众多免疫治疗手段中,嵌合抗原受体T细胞(CAR-T)疗法无疑是佼佼者。自2017年首款产品获批以来,截至2025年,全球已有14款商业化CAR-T产品上市。然而,这14款产品无一例外均属于体外制备的自体CAR-T细胞疗法。传统的自体CAR-T疗法面临诸多难以逾越的鸿沟:制备工艺流程漫长、技术门槛极高、单剂治疗成本往往高达数十万美元,且患者必须接受清淋预处理以腾出细胞扩增所需的免疫空间。为了降低成本和缩短制备周期,异体CAR-T疗法被寄予厚望,但随之而来的移植物抗宿主病(GVHD)和宿主抗移植物反应(HVGR)成为新的核心障碍——前者引发患者多器官损伤,后者则大幅缩短了异体CAR-T细胞在体内的存活时间。在此背景下,“体内CAR-T细胞疗法”作为一种颠覆性的创新替代方案应运而生。其核心思路是绕过体外复杂的细胞培养与基因工程改造环节,直接通过基因递送载体将CAR基因导入患者体内的内源性T细胞,在原位生成具有肿瘤杀伤活性的CAR-T细胞。这种“即用型”的免疫治疗方案无需清淋预处理,极大地简化了制造过程,有望将CAR-T疗法转化为适用于广泛患者群体的标准治疗手段。而实现这一宏大愿景的关键,在于选择可靠、高效且安全的基因递送载体。本文将系统综述体内CAR-T疗法的技术框架,重点聚焦病毒与非病毒两种递送载体的临床前研究进展,并深入探讨当前面临的挑战与未来发展方向。一、体内CAR-T细胞疗法的基因递送载体概况
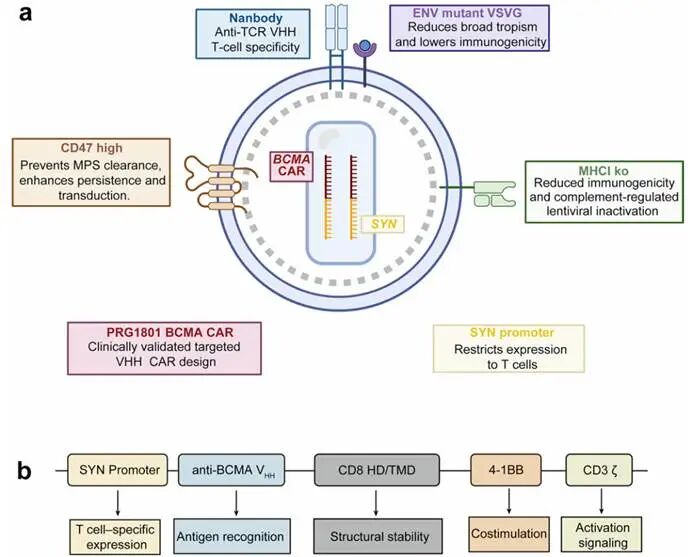
自2017年体内CAR-T核心概念确立以来,该领域进入了快速发展阶段。为了满足体内原位生成CAR-T细胞的应用要求,理想的递送载体必须具备以下特质:足够的基因承载能力以装载完整的CAR序列;高效的靶向转导效率以实现特异性T细胞修饰;较低的体内免疫原性以提升生物安全性;以及较低的制备难度以控制工业化生产成本。根据载体性质的不同,目前用于体内CAR-T构建的基因递送载体主要分为两大阵营:病毒载体与非病毒载体。病毒载体主要包括慢病毒载体和腺相关病毒载体,它们是目前CAR-T细胞治疗中最成熟的基因递送技术,凭借极高的转导率成为体内基因递送的先锋工具。非病毒载体则主要包括脂质纳米颗粒、阳离子聚合物和外泌体。相较于病毒载体,非病毒载体具有细胞毒性低、免疫原性弱以及几乎无插入突变风险的显著优势,正逐渐成为下一代制备体内CAR-T细胞的可靠替代载体。ESO-T01作为一种复制缺陷型的慢病毒载体,其设计融合了多项前沿生物工程技术,旨在实现高效、特异且安全的体内T细胞编程。首先,在特异性靶向与免疫屏蔽方面,ESO-T01对水泡性口炎病毒糖蛋白G(VSV-G)的关键残基进行了突变,消除了其广泛的哺乳动物细胞嗜性;同时,载体膜表面整合了抗T细胞受体(TCR)纳米抗体,实现了对T细胞的精准靶向;此外,载体过表达CD47,通过抑制单核吞噬细胞系统的清除作用,延长了载体在体内的循环时间,提高了转导效率。其次,在降低免疫原性方面,载体敲除了主要组织相容性复合体I类分子(MHC-I),以降低机体对载体的免疫排斥反应,从而减少潜在的炎症风险并延长CAR-T细胞的存续时间。最后,在CAR结构设计上,ESO-T01编码的人源化抗BCMA CAR包含抗BCMA单域抗体(VHH)、人CD8铰链和跨膜区、4-1BB共刺激域以及CD3ζ激活域,这一结构设计旨在平衡抗肿瘤活性与持久性。图1. ESO-T01的载体设计与CAR构建二、基于病毒载体的体内CAR-T递送系统
2.1慢病毒载体(LV)
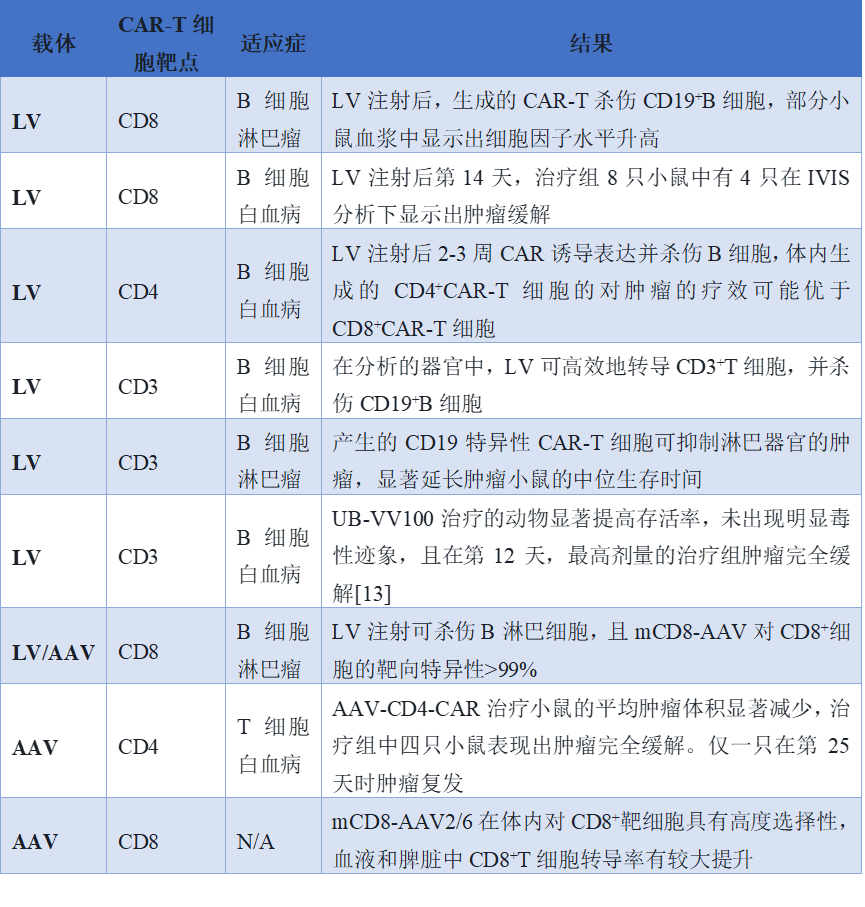
慢病毒是CAR-T细胞治疗领域应用最广泛的转基因载体。在体内CAR-T的临床前研究中,Pfeiffer团队率先证明了将特异性识别CD8的慢病毒递送至小鼠体内,能够实现对体内T细胞的原位转导,并成功表达CD19-CAR。然而,传统的慢病毒载体缺乏特异性细胞靶向能力,容易导致体内广泛的非靶向转导,存在较大的安全隐患。为了解决这一痛点,研究人员通过对病毒包膜蛋白进行工程化“假型化”改造,调节慢病毒对宿主细胞的嗜性。目前主要通过三种病毒的包膜糖蛋白进行改造:(1)囊泡性口炎病毒糖蛋白(VSV-G)。VSV-G具有优异的跨物种转导能力,但自然嗜性过广。目前的策略是保留其介导膜融合和提升颗粒稳定性的核心区域,对其进行突变改造。例如,突变型VSVGmut可与单链抗体片段构建嵌合包膜,在小鼠体内成功生成特异性CAR-T细胞;也有研究利用其改造出UB-VV100颗粒,在体内高效杀伤恶性B细胞且无明显毒性。(2)副黏液病毒包膜蛋白(以尼巴病毒NiV为主)。NiV的融合糖蛋白和附着糖蛋白能够精准识别宿主细胞表面受体并触发膜融合。经过改进的靶向NiV包膜系统与第四代慢病毒载体结合,展现出了比VSV-G伪型载体更高的T细胞特异性转导效率、更优的安全性及载体稳定性。(3)辛德毕斯病毒(SINV)结构蛋白。SINV的E2糖蛋白是决定细胞嗜性的主要因子。研究人员通过突变E2糖蛋白,使其丧失对体内非T细胞的嗜性,从而显著提升了慢病毒对T细胞的转导特异性与效率。2.2腺相关病毒载体(AAV)
腺相关病毒是一种直径约25 nm的无包膜病毒,以其持续的转基因表达能力和极低的毒性在基因治疗领域广受青睐。目前发现的AAV自然血清型超过100种,其中AAV6在T细胞转导中较为常用。Nawaz等人率先构建了携带特异性识别CD4的CAR编码基因的AAV载体,注入小鼠体内后成功转导出足够的CAR-T细胞,实现了抗肿瘤免疫效果。然而,天然AAV血清型存在固有的局限性,尤其是缺乏能高效转导T淋巴细胞的专属血清型。因此,衣壳蛋白改造成为突破的核心。例如,Adrian等人研发的AAV-T1和AAV-T2新型衣壳,对T细胞的靶向效果提升了5倍,使得所需载体剂量大幅降低;Demircan等人则将特异性识别CD8的DARPin分子插入AAV衣壳蛋白特定环区,在免疫功能正常的小鼠中实现了对CD8+ T细胞的高度特异性靶向。不过,AAV载体目前仍面临包装载荷能力有限(难以携带大片段基因)以及扩大制造规模困难等挑战。表2. 基于病毒载体的体内CAR-T细胞疗法部分临床前研究三、基于非病毒载体的体内CAR-T递送系统
3.1脂质纳米颗粒(LNP)
LNP由可电离脂质、辅助脂质、聚乙二醇修饰脂质和胆固醇组成,在mRNA疫苗的成功中发挥了核心作用。当LNP表面偶联抗体或肽段等靶向配体时,能够实现向T细胞的特异性递送。在抗纤维化领域,LNP展现出巨大潜力。研究证明,通过注射载有靶向成纤维细胞活化蛋白(FAP)CAR编码mRNA的LNP,可在体内原位生成FAP-CAR-T细胞,有效减少心肌纤维化并恢复心功能。由于mRNA未整合进基因组,这种表达是短暂的。为了实现持久表达,研究人员设计了表面修饰CD3抗体的LNP递送融合基因质粒,实现了稳定转染,使治疗效果持续数十天。此外,LNP还能在体内直接生成CAR单核细胞,用于抑制肿瘤向肝脏转移。3.2阳离子聚合物
阳离子聚合物可通过静电相互作用与核酸结合形成多聚复合物。常见的如聚酰胺-胺型树枝状高分子(PAMAM)、聚乙烯亚胺(PEI)和聚β氨基酯(PBAE)。其中PBAE因结构灵活、合成简便且成本较低而备受关注。研究表明,利用基于PBAE的纳米载体将转座酶整合到载体中,可在体内快速编辑T细胞,显著延长白血病模型小鼠的生存期。同时,该类载体也可用于体内生成具有抗肿瘤活性的CAR-M1巨噬细胞。3.3外泌体
外泌体是细胞分泌的30-150 nm纳米级囊泡,作为细胞间天然的生物分子传递媒介,具有高负载能力、高稳定性、低清除率和极低免疫原性的优势。在体外,设计有抗CD3/CD28单链抗体的外泌体可无需磁珠辅助直接激活原代T细胞并递送CAR-mRNA。在体内应用方面,带有CAR配体的基因工程外泌体不仅能作为递送工具,还能促进已有CAR-T细胞的功能持久性。多项临床试验已证实了外泌体临床应用的安全性,使其成为极具前景的体内递送载体。表3. 基于非病毒载体的体内CAR-T细胞疗法在临床前研究四、体内CAR-T递送载体当前的局限性
尽管临床前研究取得了显著的成果,但无论是病毒载体还是非病毒载体,在走向临床广泛应用的道路上仍面临严峻挑战。对于病毒载体而言,首当其冲的是插入突变风险。慢病毒载体倾向于整合到高转录活性的基因及癌症相关基因中,在直接注射入体内的过程中,这种随机整合可能诱发二次肿瘤。其次,病毒载体普遍存在脱靶效应和非靶向毒性。再次,免疫原性问题限制了病毒的重复给药,导致转导效率降低和稳定性下降。最后,AAV有限的负载能力严重限制了CAR设计的灵活性。对于非病毒载体而言,最大的短板在于缺乏基因组整合能力。由其介导的基因表达往往是短暂性的,为了维持疗效必须进行重复递送。然而,重复输注LNP会诱导机体产生抗聚乙二醇(Anti-PEG)抗体,加速载体清除;阳离子聚合物的重复给药也可能引发累积毒性。此外,非病毒载体的转染效率仍有待提高,特别是其在进入细胞质之前,从内体/溶酶体腔室逃逸的能力非常有限,容易导致遗传物质被降解。药物负载能力有限以及药物释放的不均一性,也是当前亟待解决的工程技术难题。五、部分临床研究结果
5.1体内CAR-T治疗多发性骨髓瘤的研究结果
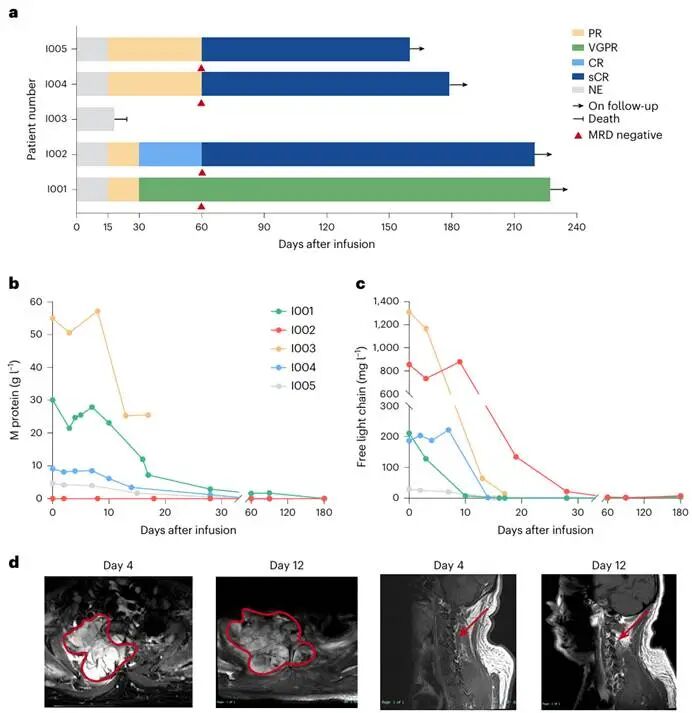
基于ESO-T01的临床试验结果表明,单次静脉输注该纳米抗体修饰的慢病毒载体即可在患者体内高效原位生成CAR-T细胞,将治疗准备时间从传统数周大幅缩短至中位约8.5小时。在疗效上,9例复发/难治性多发性骨髓瘤患者整体客观缓解率(ORR)达80%,多数患者在30-60天内达到严格完全缓解(sCR)且微小残留病(MRD)转阴,体内扩增峰值(占外周CD3+ T细胞59.1%)与持久性媲美传统体外制备CAR-T。在安全性上,该疗法呈现出独特的“双相免疫激活”特征(24小时内先天免疫波与6-14天适应性免疫波),虽引发早期一过性血细胞减少和较高比例的CRS(多为2-3级且可管可控),但通过引入预防性皮质类固醇成功实现了毒性与疗效的“解耦”。该研究不仅验证了免淋清、免体外制备的体内生成CAR-T技术的可行性与深度缓解潜力,其所展现的靶向转导与药代动力学规律,也为未来体内TCR-T等细胞治疗提供了全新的技术范式。图2. ESO-T01输注后的临床疗效及生物标志物动态变化5.2国际首个体内CAR-T治疗难治性系统性红斑狼疮(SLE)的临床研究结果
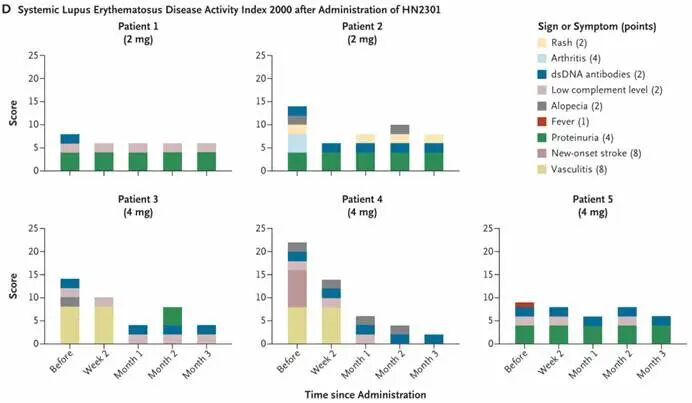
基于LNP-mRNA技术的体内CAR-T候选药物HN2301在5例难治性SLE患者中首次实现了人体内的T细胞原位重编程。研究结果显示,在免清淋、免体外制备的条件下,患者接受4mg中剂量输注后6小时内,外周血CD8+ CAR-T细胞重编程比例即可突破60%峰值,并实现循环B细胞的快速深度清零(<1个/μL);且重复给药被初步证实可行。在有效性方面,患者自身抗体显著下降,补体恢复正常,治疗3个月后SLEDAI-2K评分均实现大幅降低(最高由22分降至2分)。安全性方面,仅出现1级CRS且经单次托珠单抗即可控,无神经毒性、肝损或其他严重不良事件。该研究不仅验证了体内生成CAR-T在自免疾病中的深度清B能力与良好安全性,更提示了“即时化、一体化”的CAR-T治疗新范式在人体内取得首个临床突破。图3. 患者系统性红斑狼疮疾病活动指数(SLEDAI-2000)评分变化六、未来展望与发展思路
作为一种具有划时代意义的免疫疗法,体内CAR-T已经开始向临床试验阶段迈出坚实步伐。例如,EsoBiotec公司研发的基于慢病毒载体的体内CAR-T疗法(ESO-T01),在多发性骨髓瘤患者的早期临床研究中,低剂量给药即让部分患者实现严格完全缓解。这一里程碑式的数据验证了体内CAR-T的临床可行性。为了满足未来大规模临床应用的需求,递送载体的迭代升级将沿着以下几个核心路径展开:(1)开发融合优势的新型递送平台。例如蛋白脂质载体(PLV)整合了病毒蛋白的高效侵染与非病毒脂质体的低毒性优势,能够安全有效地完成核酸的体内递送。结合高密度脂蛋白等仿生学设计,可进一步减轻非靶向毒性。(2)联合前沿基因编辑技术。将体内CAR-T的构建与CRISPR/Cas9等精确基因编辑手段结合,不仅可以将CAR基因精准敲入安全位点,还能通过敲除免疫检查点或耗竭相关基因(如Nexin-9)来显著提升体内T细胞的抗肿瘤效能与持久性。(3)突破非病毒载体的表达时效瓶颈。针对mRNA表达时长有限的问题,环状RNA和自复制RNA是两种极具潜力的解决方案。环状RNA具有极高的稳定性和低免疫原性;而自复制RNA在临床试验中已展现出诱导快速、强效且持久免疫反应的能力,能够以更低的剂量表达更多的目的蛋白。七、结语
总的来说,体内CAR-T细胞疗法通过基因递送载体在患者体内原位“制造”杀手细胞,彻底颠覆了传统细胞疗法的生产与治疗模式。从慢病毒、腺相关病毒的假型化改造,到脂质纳米颗粒、外泌体等非病毒系统的精准化设计,递送载体的创新正在为这一领域注入源源不断的动力。尽管我们在脱靶效应、安全性控制、载药量及基因持久表达等方面仍面临诸多挑战,但随着材料科学、病毒学、免疫学与基因编辑技术的深度交叉融合,这些技术壁垒正在被逐一攻克。可以预见,在不久的将来,体内CAR-T疗法将成功跨越从临床前到临床应用的鸿沟,不仅为晚期肿瘤患者提供一种真正“即用型”、低成本且高效的安全治疗方案,更有望在自身免疫性疾病、纤维化甚至抗衰老等领域大放异彩,开启免疫治疗的新局面。[1] Xu J, Liu L, Parone P, Xie W, Sun C, Chen Z, Zhang J, Li C, Hu Y, Mei H. In-vivo B-cell maturation antigen CAR T-cell therapy for relapsed or refractory multiple myeloma. Lancet. 2025 Jul 19;406(10500):228-231. doi: 10.1016/S0140-6736(25)01030-X.[2] An N, Wang D, Zhang P, Zhang J, Parone P, Hu J, Bao Y, Xu L, Ruan H, Wan Y, Wen X, Gao Y, Li C. In vivo generation of anti-BCMA CAR-T cells in relapsed or refractory multiple myeloma: a phase 1 study. Nat Med. 2026 Mar 25. doi: 10.1038/s41591-026-04244-6.[3] Wang Q, Xiao ZX, Zheng X, Wang G, Yang L, Shi L, Xiang N, Wang X, Zha GF, Schett G, Chen Z. In Vivo CD19 CAR T-Cell Therapy for Refractory Systemic Lupus Erythematosus. N Engl J Med. 2025 Oct 16;393(15):1542-1544. doi: 10.1056/NEJMc2509522.[4] Agarwal S, Hanauer JDS, Frank AM, Riechert V, Thalheimer FB, Buchholz CJ. In Vivo Generation of CAR T Cells Selectively in Human CD4+ Lymphocytes. Mol Ther. 2020 Aug 5;28(8):1783-1794. doi: 10.1016/j.ymthe.2020.05.005.[5] Mukalel AJ, Hamilton AG, Billingsley MM, Li J, Thatte AS, Han X, Safford HC, Padilla MS, Papp T, Parhiz H, Weissman D, Mitchell MJ. Oxidized mRNA Lipid Nanoparticles for In Situ Chimeric Antigen Receptor Monocyte Engineering. Adv Funct Mater. 2024 Jul 3;34(27):2312038. doi: 10.1002/adfm.202312038.[6] Zhang Z, Ma B, Li B, Li Z, Gao M, Zhao H, Peng R, Hu J, Wang Y, You W, Gui X, Wang R, Hu X, Chen B, Zhang Y, Hao Y, Sun X, Rao P, Zhang L, Lu M, Zhou D, Yang Y, Deng M, Miao L. Cardiolipin-mimic lipid nanoparticles without antibody modification delivered senolytic in vivo CAR-T therapy for inflamm-aging. Cell Rep Med. 2025 Jul 15;6(7):102209. doi: 10.1016/j.xcrm.2025.102209.
转载说明:本文系转载内容,版权归原作者及原出处所有。转载目的在于传递更多行业信息,文章观点仅代表原作者本人,与本平台立场无关。若涉及作品版权问题,请原作者或相关权利人及时与本平台联系,我们将在第一时间核实后移除相关内容。
 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库