 五度妙笔
五度妙笔 企业透视镜
企业透视镜 API商城
API商城
 数据库
数据库8死76伤!FDA勒令撤市,安进拒绝低头


近日,美国FDA药品评价与研究中心(CDER)以“有效性从未被有效证明”和“上市申请包含不实陈述”为由,正式提议撤销将安进(Amgen)旗下罕见病药物Tavneos(通用名阿伐可泮)的上市许可。一款上市不到五年的罕见病口服药,以如此方式走到被“逐出市场”。早在今年2月,FDA便已要求安进将Tavneos撤出市场,但遭到安进拒绝。眼下,美国监管机构正在进一步施压。

Tavneos是一种全球首创的口服补体5a受体(C5aR)拮抗剂,由ChemoCentryx公司原研,于2021年9月率先在日本获批,同年10月获得美国FDA批准,成为十年来首个获批治疗ANCA相关血管炎的新药。2022年1月,Tavneos获得欧盟委员会批准上市,同年10月,安进以约37亿美元收购ChemoCentryx,将Tavneos纳入其炎症管线。此后,Tavneos相继在澳大利亚、加拿大等多个国家获得许可,并于2024年10月在中国通过NMPA批准上市。

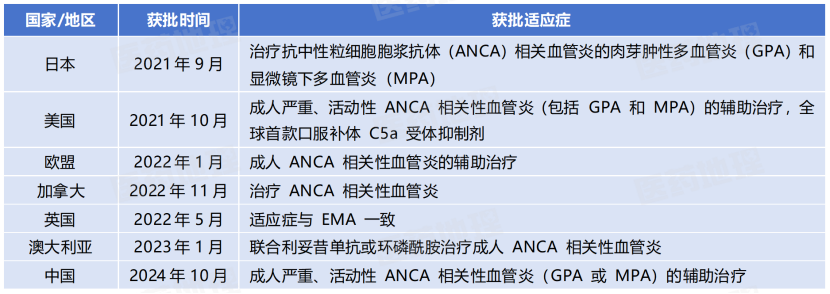
Tavneos获批历程
来源:Pharma ONE药物研发大数据平台,中国医药工业信息中心
点击“阅读原文”获取更多数据

FDA的“数据操纵”指控

就在3月底,FDA发布了措辞严厉的公告,指出截至2024年10月,Tavneos已出现76例严重药物性肝损伤(DILI)病例,其中包括54例住院和8例死亡。共有60例提供了可确定初始肝损伤模式的实验室信息;其中大多数(38例)为胆汁淤积型或混合型,常伴有碱性磷酸酶和总胆红素显著升高。共有73例提供了从开始使用阿伐可泮至DILI发病的时间,中位发病时间为46天(范围22至140天)。大多数病例(66例)来自日本,其次为美国(5例)、欧洲(4例)和加拿大(1例)。
有7例报告了经活检证实的胆管消失综合征(VBDS)为DILI的并发症,且有合理证据表明与使用Tavneos存在因果关系。所有病例均报告住院(7例),其中3例致死。初始肝损伤模式为:4例为胆汁淤积型或混合型,3例为肝细胞型。这7例病例从开始使用Tavneos到DILI发病的中位时间为46天(范围33至59天)。病例来自日本(6例)和加拿大(1例)。


然而,单凭上市后安全问题很难直接触发撤市。真正将Tavneos推向悬崖边缘的,是更深层的“数据操纵”指控。
CDER指控非盲态研究人员对关键III期ADVOCATE试验的主要终点结果进行了“操纵”:当原始分析无法证明药物有效时,他们对331名患者中9名受试者的终点进行了重新裁定,使原本不具统计学显著性的结果“看起来有效”,而原始分析甚至从未被提交给FDA。CDER表示,如果原始分析如期呈报,这项研究根本不会被视为有效性证据。
换句话说,FDA不只是在说“这款药现在很危险”,而是在说“这款药从来就不该获得批准”。
仅4.6亿的产品,为什么值得死扛?
以安进2025年全年351亿美元的总盘子来看,Tavneos不过贡献了4.6亿美元——占比仅为1.3%。单纯从财务损益的角度,安进没有为这点营收与两大监管机构正面对抗的充分理由。
2022年,安进斥资约37亿美元收购ChemoCentryx,Tavneos便是这笔交易的皇冠明珠。如今安进若在FDA和EMA的联合压力下“自愿撤市”,意味着从当前的财政损失到此前投入的数十亿美元几乎血本无归。此外,Tavneos还是安进增速最快的产品之一,2025年同比增长高达62%。安进高管反复强调,基于多年的临床数据和真实世界证据,它是一款“重要且有效的药物”。

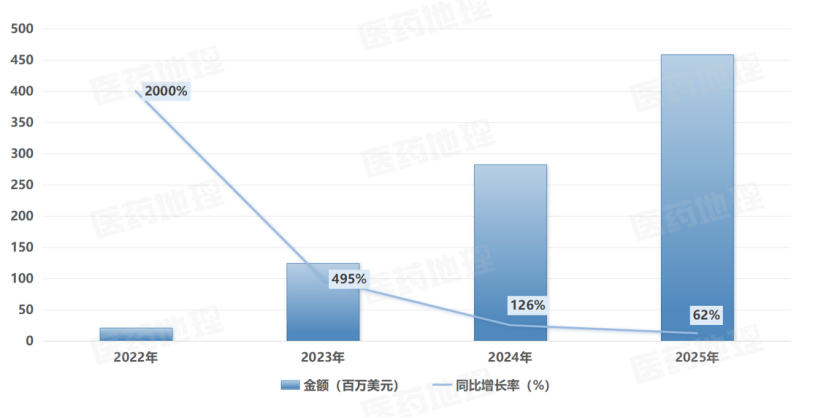
Tavneos近年全球销售额 来源:药企财报
更重要的是,当下安进一旦接受“批准时的注册研究数据经不起事后审视”这一监管定论,其整个产品管线都将被投下阴影。死扛Tavneos,守的不只是4个亿的年销售额,更是安进整个并购扩张战略的信用根基。
横跨60年的FDA临床试验改革
与Tavneos的“事后算账”形成鲜明对照,4月28日,FDA宣布将改革 60 年未变的临床试验模式。核心是:在AI和先进数据科学的驱动下,允许FDA科学家在临床试验进行期间实时查看安全信号和终点数据,而非等待数据收集完毕、完成分析后再行提交。此番作为无疑可以减少“数据透明度缺失”,让监管者能够“看见”数据的产生而非只“验收”数据的结论,从而压缩人为操纵的空间。

FDA还表示,当前一款新药从研发到获批通常需10到12年,其中近45%的时间消耗在文书工作上,这种“死时间”严重拖慢了创新疗法惠及患者的速度。新模式下,FDA审评人员可通过云端平台同步看到受试者是否出现发热等不良事件,或在影像学判读时即观察到肿瘤缩小,从而有望在试验尚未完成时就做出监管决策。
阿斯利康正在其BTK抑制剂Calquence联合疗法的II期Traverse试验中应用该模式,相关数据已通过Paradigm Health平台提交并获验证;安进则将其DLL3 T细胞衔接器Imdelltra的Ib期试验纳入试点,该研究正处于中心筛选尾声。
在隐私保护方面,FDA表示,机构无意获取患者个体数据,实时审评范围仅限于事先商定的安全性和有效性终点,初期仍将与传统申报数据并行使用,试点结束后评估改进方向。
至于安进与FDA之间的下一步——根据程序,安进可以选择正式要求听证,FDA局长将决定是否举行公开听证,而后再就撤市做出终裁。这注定是一场漫长的对峙。但无论结局如何,这款药的故事已经给整个制药行业留下了一道无法回避的考题:当药物安全与商业利益正面交锋,那些无辜的患者,他们的生命又由谁来守护?








