 五度妙笔
五度妙笔 API商城
API商城
 数据库
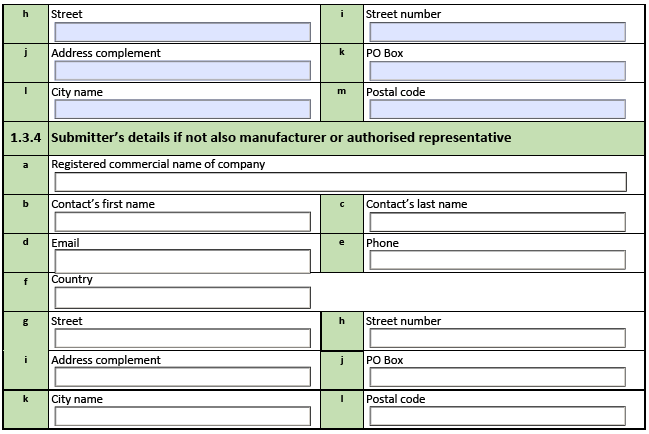
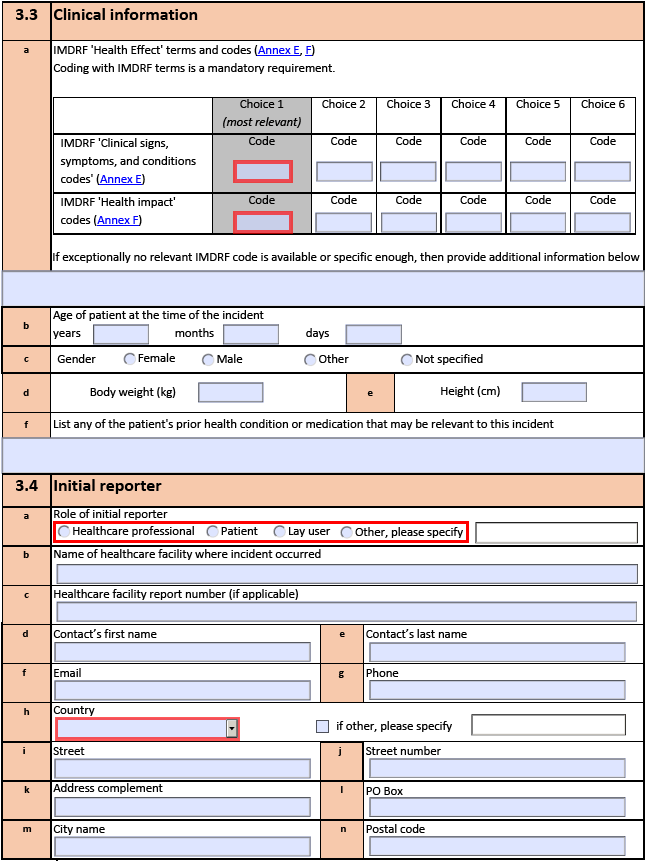
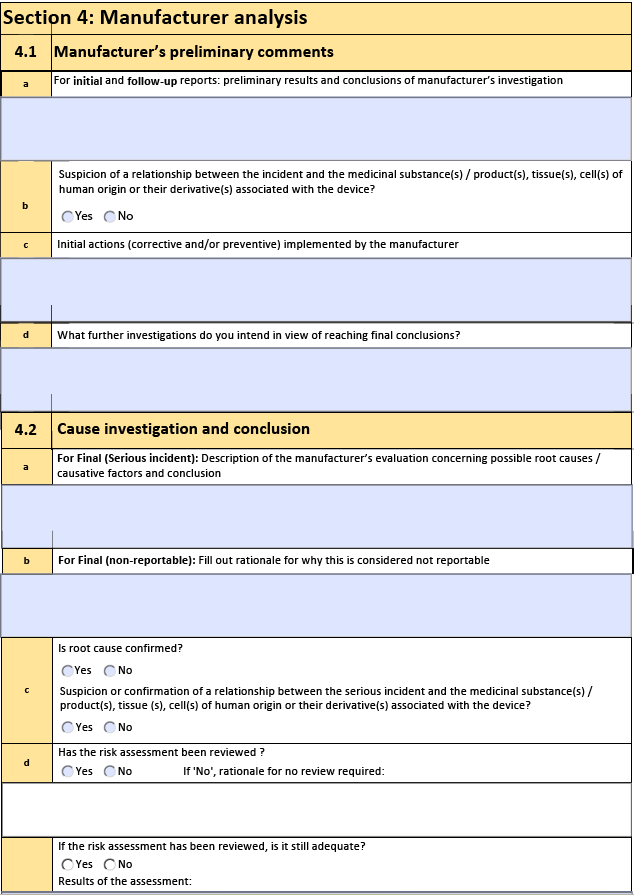
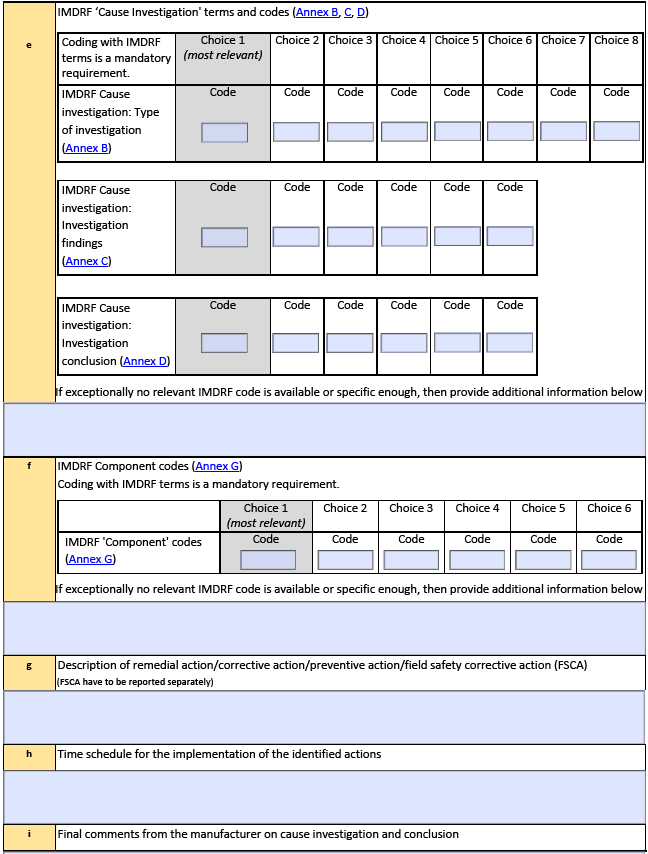
数据库欧盟新版制造商事故报告MIR,5月1日起强制执行(附模板)
欧盟网站发布: 全新版本制造商事故报告MIR表格 ,并要求从2026年5月1日起强制实施。
什么是MIR?
英文全称:Manufacturer Incident Report,中文名:制造商事故报告,由欧盟委员会制定的一种标准化报告工具,供制造商向相关主管当局报告其产品于欧盟发生的严重事故。
MIR 表格的更新源自《医疗器械法规MDR》和《体外诊断医疗器械法规IVDR》的新要求,包括涵盖"遗留器械"要求。
哪里可以找到新版 MIR 表格?
新版表格原文↘





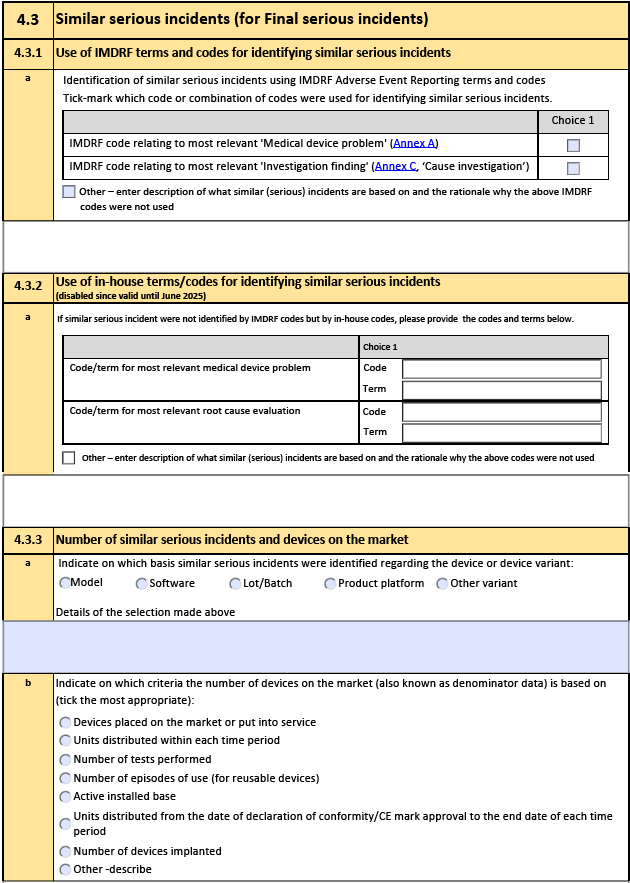
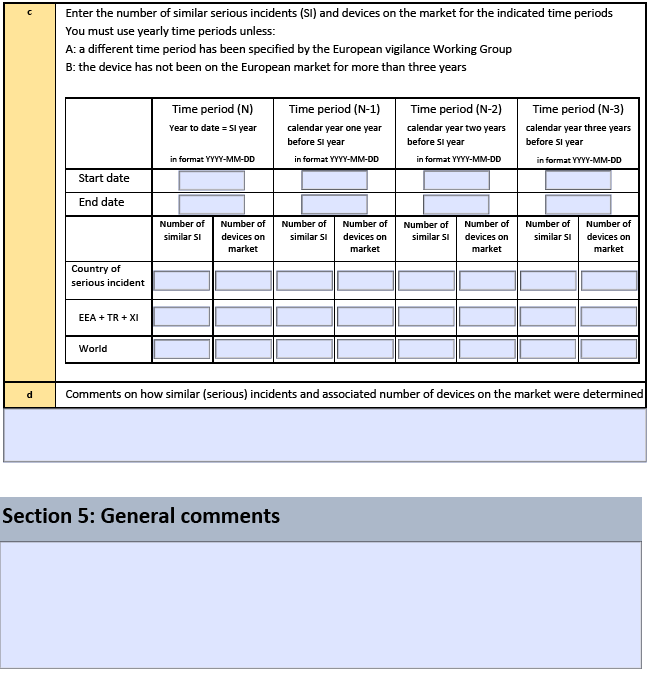

以上网址中还提供以下表格及模板:
• Field safety corrective action - FSCA 现场安全纠正措施
• Field safety notice template 现场安全通知模板
• FSN customer reply 客户回复
• FSN distributor/importer reply 经销商/进口商回复
• FSN Q&A 问与答
• Trend report 趋势报告
• Periodic summary report 定期总结报告
相关知识:
2025年12月,欧盟医疗器械协调小组(MDCG)正式发布 MDCG 2025-10 指南文件,对医疗器械(MDR)与体外诊断医疗器械(IVDR)的上市后监督(PMS) 进行系统、全面的阐述。该文件并非新增法规要求,而是对 MDR/IVDR 中 PMS 相关条款的权威解读与实操指引。
一、 为什么PMS如此重要?
1.首先明确PMS在欧盟法规体系中的核心地位:
•PMS 是 质量管理体系(QMS)不可分割的一部分
•覆盖器械整个生命周期
•目的是确保器械在真实世界使用中的安全性、性能与获益-风险比持续可接受
2.法规明确要求制造商:
•主动、系统性 收集上市后数据
•利用数据持续更新:风险管理、临床/性能评价、技术文档
•必要时采取 CAPA 或 FSCA
3.PMS不再是“被动处理投诉”,而是一个持续运行的闭环系统。
二、适用范围与目标
1.适用范围:
•适用于所有医疗器械(MD)和体外诊断器械(IVD)
•包括所有风险等级
•涵盖定制器械
2.指南目标:
·解释什么是 PMS 系统
·说明PMS 计划的构成
·描述PMS 的主要活动
·阐明PMS 与 QMS 其他模块的 联动关系
3.明确不涵盖:
•PSUR / PMS Report 的具体写作模板(已有其他 MDCG 指南)
•医疗机构自制器械(Article 5(5))
三、法规要求下的PMS系统
1. PMS 的基本要求
制造商必须:
•规划、建立、实施、维护并持续更新 PMS 系统
•PMS 的深度与复杂度必须 与器械风险等级和类型相匹配
PMS 系统应当能够:
•主动收集真实世界数据
•分析质量、性能与安全性
•支持是否需要采取行动的决策
2. PMS 与 QMS 的关系
指南强调:
•PMS 是 QMS 的输入源
•PMS 数据必须反馈至:风险管理、临床 / 性能评价、设计变更、标签和说明书、CAPA 与 FSCA
未深度融合QMS的PMS将被视为不合规。





