 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库全是考点|欧盟MDR CE质量体系的公告机构必审内容
欧盟MDR法规对制造商的质量管理体系QMS,提出了相较原指令MDD/AIMDD 更为系统化、文件化、深度融合临床安全和合规性要求 。本期将解读聚焦:申请CE证书制造商QMS的MDR核心要求。
一. MDR下QMS的强制性法律基础
MDR 第10条第8款规定: 制造商必须建立/文件化/实施/维护/持续改进并定期更新一个能够确保器械符合MDR法规全部要求的质量管理体系(QMS) 。该QMS规定必须至少覆盖以下法定内容:
1. 法规符合性策略( 含符合性评估途径选择);
2. 设计控制与设计转移 (设计变更需重新评估法规符合性);
3. 风险管理 (贯穿全生命周期且与临床证据闭环);
4. 生产过程控制/最终检验及验证 ;
5. 供应商与外包方控制 (尤其是关键工序/灭菌/软件服务商);
6. 上市后监督系统 (含上市后临床跟踪PMCF计划);
7. 抱怨处理与纠正/预防措施CAPA ;
8. 质量管理体系内部审核 ;
9. 人员培训与能力管理 ;
10. 与主管当局、公告机构、经济运营者(进口商/分销商)的接口流程 。
二. 对比旧指令QMS的MDR增量要求
1. 法规符合性负责人PRRC的指定
QMS要求制造商必须明确指定1名员工为PRRC(该角色也可选择外包,但需证明可控性),要求此人具备特定资质(法律/医学/工程+法规经验),职责涉及:监督体系运行、技术文档维护、上市后监督PMS报告、警戒系统等。
2. 风险管理与临床评估的关系
MDR法规要求,风险管理输出直接驱动临床评估计划、临床评价、上市后临床跟踪PMCF。
QMS需确保:每个已识别风险所对应的风险控制措施,均需被临床数据验证(若必要);上市后监督所收集的现场信息必须触发风险-受益的再评估。
3. 上市后监督作为质量管理体系的核心子系统
PMS不再停留于“处理抱怨”,而是涵盖:
· PMS计划 (数据收集方法、指标等);
· PMS报告 (I类器械)或 PSUR定期安全更新报告 (IIa/IIb/III类,至少每年更新1次),PSUR必须包括风险-受益结论、销售/使用量、不良事件趋势、PMCF进展等。
· QMS需确保上市后监督数据能够反馈至设计变更、风险管理、临床评价。
4. 全生命周期内对变更的控制
所有可能影响安全或性能的变更(如材料、工艺、预期用途、软件算法),都必须在质量管理体系QMS中进行触发,例如:
· 变更评估流程 (是否需新临床数据或公告机构评审);
· 变更后的符合性声明更新;
· 通知NB公告机构 (依据MDR Annex VII, 4.10.2)。
5. 与经济运营商的流程对接
制造商必须在质量管理体系中予以明确以下问题:
· 如何向欧盟授权代表提供完整的技术文档?
· 怎样确保进口商/分销商能够接收到上市后监督和现场安全纠正措施FSCA信息?
· 供应链的可追溯性(UDI载体与记录)。
三. MDR公告机构对QMS的审查重点
公告机构在MDR CE审核时,依据Annex IX(基于QMS的符合性评估)实施评审,其关注重点如下:
· QMS与MDR法定要素的对应表 (通常要求开展GAP分析);
· PRRC的任命、职责证据;
· PMS/PSUR流程及实际输出样本;
· 临床评价与风险管理的联动记录 (如风险是否被临床数据充分覆盖);
· 变更管理流程的触发实例;
· 供应商控制 (尤其是关键件、灭菌、软件外包)。
注意:针对IIa类及以上器械,通常结合技术文档评审同时开展QMS现场审核。
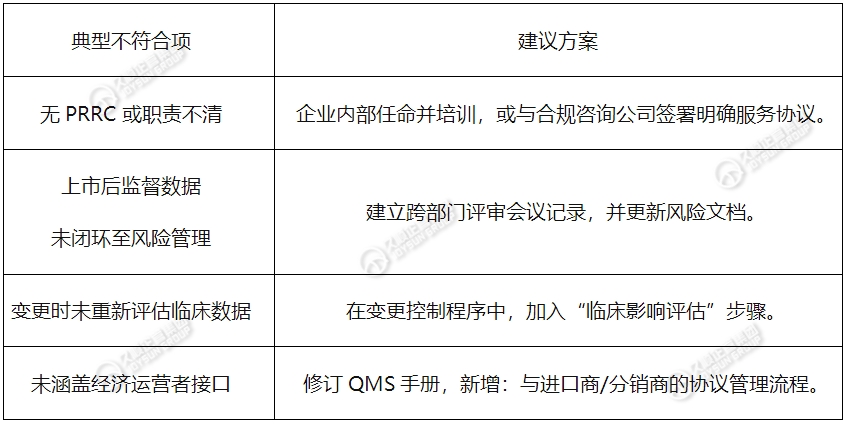
四. MDR体系审核中典型不符合项及应对
五. 总 结
MDR法规要求制造商的质量管理体系不再仅是“质量保证”,而是 法规符合性、风险管理、临床证据、上市后监控的四位一体动态系统 。
制造商必须在申请CE证书前,完成现有QMS与MDR的差距分析,并将PRRC、PMS/PSUR、变更的临床影响评估等作为强制模块嵌入。
建议:制造商在将CE申请提交公告机构前,先至少开展1轮内部模拟审核,*重点验证→ PMS 到 CAPA 再到 设计变更 的端到端闭环能力 。





