 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库QC数据可靠性检查新规下,色谱软件合规精准化
2026年5月7日,国家药品监督管理局食品药品审核查验中心(CFDI)发布《药品质量控制实验室数据可靠性检查指南(征求意见稿)》(以下简称“《指南》”)。《指南》以ALCOA++为基本原则,围绕数据管理、计算机化系统检查要点、常见缺陷和检查示例建立了较为系统的检查框架,并在附件中提供色谱、理化、微生物限度、中药指纹图谱等检验方法的数据可靠性检查清单。
与以往较多停留在原则层面的要求相比,《指南》对QC实验室日常工作中“怎么查、查什么、哪些环节容易忽略”给出了更具操作性的提示。特别是在色谱系统场景下,《指南》不仅关注数据是否被保存,更关注数据从样品接收、序列建立、进样采集、积分处理、结果审核、报告输出到归档备份的全过程是否满足ALCOA++原则。
识林数据库统计显示,2015年至2025年间,FDA就因色谱数据可靠性问题发出98封警告信;在国内GMP检查中,计算机化系统的账户权限、审计跟踪、数据备份等问题也仍是高频缺陷项。对于QC实验室而言,色谱系统既是检验数据产生、处理、审核和报告的核心工具,也是数据可靠性风险最集中、最容易被检查深入追溯的系统之一。
识林在2025年联合华谱科仪编写了《色谱软件的数据可靠性保障与电子记录合规策略》白皮书(以下简称“《白皮书》”)。借新规发布之际,本文尝试结合《白皮书》,将监管要求转化为系统、合规、可落地的系统能力,供识林读者参考。
一、指南对色谱系统的数据可靠性要求
《指南》对色谱系统的要求可以概括为一个核心逻辑:数据可靠性不是单一软件功能,而是“系统设计、权限控制、数据生命周期、审计跟踪、验证确认和日常管理”共同形成的受控能力。企业在理解和落实相关要求时,可重点关注以下六个方面。
1. 原始数据与元数据应形成完整链条
色谱原始数据、结果数据、方法、序列、积分参数、系统消息、审计跟踪等均应被纳入管理。导出的PDF或纸质报告通常只是副本,不能替代系统中的原始电子记录。
2. 账户权限应支持“单人单号”和最小权限
系统应避免共享账户和匿名操作,按岗位职责设定角色权限。特别是方法修改、序列编辑、数据删除、审计跟踪配置、管理员权限等高风险操作,应被严格限制并留痕。
3. 审计跟踪不仅要“有”,还要可审核
审计跟踪应覆盖数据创建、修改、删除、处理和关键配置变化,并能反映“何人、何时、何事、为何”。企业还需要建立审计跟踪审核机制,避免系统记录存在但未被有效使用。
4. 积分处理和异常数据应被技术与程序双重控制
手动积分、重新处理、中止进样、失败运行、异常序列等是色谱数据可靠性检查中的重点。系统应支持保留自动积分与手动积分版本、记录变更理由,并通过第二人复核或批准形成控制闭环。
5. 备份、恢复与归档要能证明数据长期可用
数据备份不只是IT任务,还应满足GMP数据管理要求。企业需要关注备份介质、异地保存、恢复测试、归档后可读性、元数据和审计跟踪同步保存等问题。
6. 计算机化系统验证应覆盖预期用途和关键风险
色谱软件的合规性不能仅看供应商功能清单,还应结合企业实际使用场景开展URS、风险评估、确认与验证、权限测试、审计跟踪测试、备份恢复测试及周期性回顾。
二、《指南》与《白皮书》:从法规要求到设计实务
从专业参考价值看,《白皮书》一方面梳理FDA、EMA、MHRA、WHO、PIC/S、NMPA等机构对数据可靠性、电子记录、电子签名、计算机化系统验证的要求;另一方面结合色谱数据管理的实际流程,讨论采集与处理、审核与报告、存储与归档、备份与恢复、账户权限、审计跟踪、电子签名和CSV等关键环节,结合行业专家的观点,展开谈论。
《白皮书》尝试回答企业在色谱系统合规管理中经常遇到的几个问题:监管要求如何对应到软件设计?哪些系统配置会影响数据可靠性?哪些商业模式或使用习惯可能诱发共享账户、权限过度、备份失控等“无意的”合规风险?如何把警告信中的缺陷经验转化为系统选型、验证和日常管理的检查点?
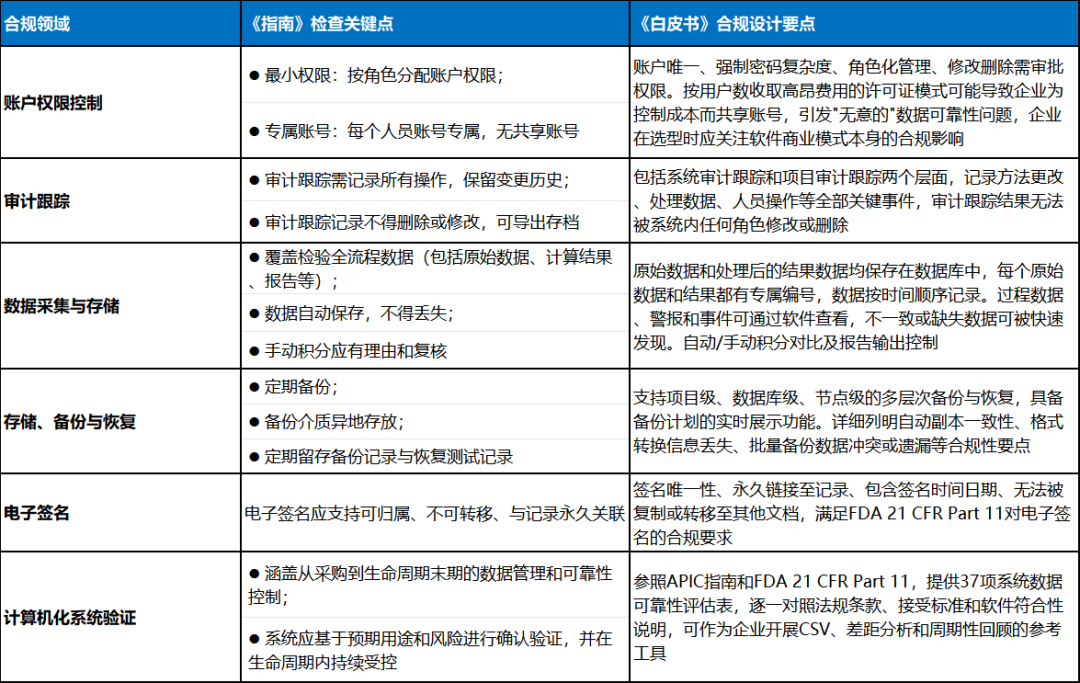
《指南》要求与《白皮书》参考价值对照
此外,《白皮书》还吸收了中外专家的评审意见。前WHO与MHRA检查员、WHO数据可靠性指南主要起草人Ian Thrussell先生,从国际检查视角提出数据加密存储、多时区管理、SaaS商业模式对合规的影响等问题;国内制药行业资深IT专家史俊先生,则从企业信息化实践角度关注系统与LIMS/QMS的对接、备份—恢复闭环机制和V型验证模型等议题。
这些讨论的价值在于提醒企业:色谱软件合规不能停留在“有没有审计跟踪、有没有电子签名、能不能导出报告”的功能层面,而要回到系统如何被配置、验证、使用、审核和维护。只有当技术功能、管理程序、人员职责和质量文化形成闭环,软件功能才可能转化为稳定可靠的合规能力。
三、企业可以做些什么
《指南》的发布,意味着QC实验室数据可靠性检查正在进一步标准化、清单化和场景化。色谱分析作为药品质量控制的核心环节,其电子数据管理能力将直接影响企业对检验结果真实性、完整性和可追溯性的证明能力。数据可靠性检查将更加标准化、精细化和常态化。
除了熟读《指南》并提出充分的且建设性的反馈意见,对于制药企业而言,以下行动项值得考虑:
对照《指南》附件中的色谱数据可靠性检查清单,开展QC实验室自查,特别关注权限、审计跟踪、积分处理、异常运行、备份恢复等“易忽略”环节。
以色谱系统数据可靠性评估表为基础,对现有CDS或色谱软件开展差距分析,明确哪些问题属于系统功能缺口,哪些属于配置、验证或SOP执行问题。
在新系统选型或老系统升级时,将数据可靠性要求前置到URS和供应商评估中,避免系统上线后再补救权限、审计跟踪、备份恢复和电子签名等基础控制。
建立跨部门管理机制,由QC、QA、IT和供应商共同参与系统验证、权限管理、审计跟踪审核、备份恢复测试和周期性回顾。
识林会员也可点击文末链接阅读《白皮书》,以全球法规要求为基础,以色谱数据管理流程为主线,结合警告信分析、专家意见和系统评估工具,深入理解新规要求,并系统性评估现有色谱系统、完善电子记录管理和计算机化系统验证。
参考资料:
1. CFDI《药品质量控制实验室数据可靠性检查指南(征求意见稿)》,2026年5月7日发布。
2. 华谱科仪、识林:《色谱软件的数据可靠性保障与电子记录合规策略》白皮书。
|责任编辑:识林-木姜子
|编辑:识林-雪见
识林®版权所有,未经许可不得转载

【关于识林-常见问题与解答】






