 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库周末文摘 | 人工智能在药品监管应用中的法律挑战探讨

引用本文



王翔宇,侯艳,杨蕾,王海南,袁林*.人工智能在药品监管应用中的法律挑战探讨[J].中国食品药品监管.2026.04(267):4-15.
人工智能在药品监管应用中的法律挑战探讨
Legal Challenges of Artificial Intelligence in Drug Regulation
王翔宇
国家药品监督管理局科技和国际合作司
WANG Xiang-yu
Department of Science, Technology and International Cooperation, National Medical Products Administration
侯艳
北京大学
HOU Yan
Peking University
杨蕾
北京大学
YANG Lei
Peking University
王海南
国家药品监督管理局药品监督管理司
WANG Hai-nan
Department of Drug Supervision, National Medical Products Administration
袁林*
中国药品监督管理研究会
YUAN Lin*
China Society for Drug Regulation
摘 要Abstract
本文聚焦人工智能应用于药品监管领域的法律问题研究,通过系统梳理欧盟、美国、英国、日本等主要法域在数据安全、监管责任、数据利用边界、隐私保护、知识产权及商业秘密等方面的制度进展,分析人工智能嵌入药品监管流程后对监管规则和治理机制提出的新要求。基于对主要法域制度安排和实践路径的比较研究,本文总结了国际监管在高风险治理、数据治理、人类监督、生命周期管理和透明义务等方面的趋同趋势,进而提出对我国药品监管制度构建的启示,旨在为统筹技术创新、监管效能与公众安全提供政策参考。
This study examines the legal issues arising from the application of artificial intelligence (AI) in drug regulation.Through a systematic review of regulatory developments in major jurisdictions, including the European Union, the United States, the United Kingdom, and Japan, it analyzes key institutional progress across the following dimensions: data security,regulatory liability, data governance boundaries, privacy protection, intellectual property, and trade secrets. The study further explores the new requirements imposed on regulatory rules and governance mechanisms following the integration of AI into drug regulatory processes. Based on a comparative analysis of institutional arrangements and practical approaches across jurisdictions, it identifies a growing convergence in key areas, including high-risk governance, data governance, human oversight, lifecycle management, and transparency obligations. On this basis, the paper discusses implications for the development of China’s regulatory framework for AI in drug regulation, aiming to provide policy insights for balancing technological innovation, regulatory effectiveness, and public safety.
关键词Key words
人工智能;药品监管;数据安全;监管责任
artificial intelligence; drug regulation; data security; regulatory liability
基金项目



中国食品药品国际交流中心项目

2018 年以来, 以预训练大模型为代表的新一代人工智能(artificial intelligence,AI)快速演进,推动AI 由感知智能向认知智能拓展、由判别式应用向生成式应用深化,并逐渐成为生物医药产业升级和监管体系数字化转型的重要支撑。面对数据规模持续扩张、监管任务日益复杂和风险形态不断变化等情况,我国药品监管部门正依托《药品监管人工智能典型应用场景清单》[1]推进“人工智能+ 药品监管”实践,国家药品监督管理局(以下简称国家药监局)于2026 年4 月发布《关于“人工智能+ 药品监管”的实施意见》,对AI 在药品全生命周期监管中的创新应用作出系统部署[2],标志着我国药品监管中的AI 应用已由场景探索转向体系化推进与制度化布局。
但药品监管并非一般技术应用场景,而是兼具高敏感数据处理、高影响行政判断和高公共安全风险的特殊治理领域。监管活动涉及企业申报资料、临床试验数据、不良反应报告和内部调查信息等多类型数据,往往同时关联个人信息、商业秘密、知识产权和监管资料等多重权益;相关判断又直接影响药品上市、风险识别和监管处置,关系企业合法权益、患者生命健康与监管公信力。因此,AI 一旦进入审评、检查、药物警戒等高影响环节,其意义便不再只是提升效率,而是可能进一步介入事实识别、风险研判和决定形成过程,从而对监管活动的合法性、正当性、透明性与可归责性提出新的要求。
基于此, 本文关注的并非AI 能否用于药品监管,而是其进入监管决策链条后, 如何在法治轨道上实现可控、可追溯、可归责的使用。值得关注的是,2026 年1 月, 美国食品药品监督管理局(Food and Drug Administration,FDA)与欧洲药品管理局(European Medicines Agency,EMA) 联合发布《药品研发中良好AI 实践指导原则》(Guiding Principles for Good AI Practice in Drug Development),提出以人为本设计、风险治理、数据治理、性能评估和全生命周期管理等10 项原则[3],并在国际药品监管机构联盟(International Coalition of Medicines Regulatory Authorities,ICMRA) 框架下发起对该指导原则的全球讨论。由此可以看出,AI 在药品监管中的应用已不再只是单一法域内部的技术问题,而正在成为借助监管协调开展全球博弈的新制高点。
基于这一背景,本文以AI 辅助审评和AI 药物警戒为观察窗口,围绕AI 在药品监管中所引发的数据安全、责任分配、数据利用边界、隐私保护和知识产权等问题展开分析,重点关注欧美日英等国家和地区的制度设计和法律规定,从而在比较国际制度进展的基础上,提炼对我国药品监管制度构建的启示。下文据此对国际制度进展作概括,并尝试提炼若干具有共通意义的制度发现。
1 药品监管中AI 使用的核心法律问题
AI 辅助审评和AI 药物警戒并非两类彼此分离的法律议题,而是观察同一组宏观法律问题的两个高影响窗口。在辅助审评场景中,模型参与资料筛选、差异识别和风险提示时,往往同时牵涉企业资料控制边界、商业秘密保护、模型输出能否进入行政证据链以及由此产生的责任归属;在药物警戒场景中,模型用于信号识别、聚类分析和风险预警时,则会同步触发数据来源合法性、患者信息保护、模型可解释性和风险处置可归责性等问题。由此可见,数据安全、监管责任、数据利用边界、隐私保护、知识产权与商业秘密,并不是AI 应用中的若干并列事项,而是沿着“数据进入- 模型处理- 结果使用-决定形成”这一链条交织生成、共同作用于监管决策的合法性与可归责性。上述AI 嵌入药品监管决策链条并由此引发的主要法律问题,如图1 所示。基于这一认识,本文按5 类核心法律问题展开分析,以展示AI 嵌入药品监管流程后所触发的主要规范挑战;在后文则进一步将其归纳为责任锚定、边界控制和透明可审查3项治理抓手,以说明不同法律问题如何在制度构建层面形成可操作的统一回应。
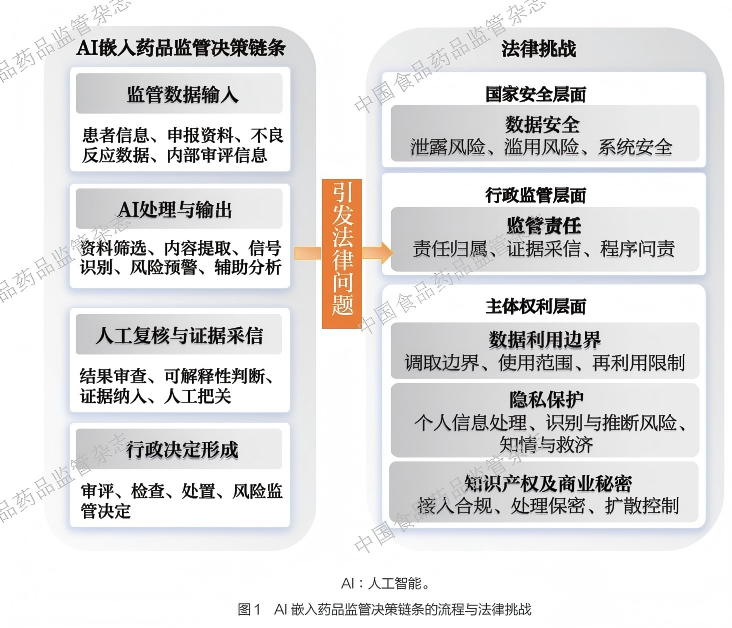
1.1 数据安全与模型可控边界
在药品监管语境中,数据安全已不再是传统意义上的信息系统防护问题,而是涵盖数据输入、模型调用、结果留存、日志审计、供应链接触和再利用限制的全过程治理问题。监管机构所处理的数据往往兼具个人信息、商业秘密、知识产权客体和非公开监管资料等多重属性,一旦脱离可控边界,其风险后果便不仅表现为数据泄露本身,还会进一步外溢至审评结论、风险预警和监管处置的合法性基础。
面对此挑战, 目前欧盟的制度安排具有体系性。《通用数据保护条例》(General Data Protection Regulation,GDPR)[4]和《欧盟机构数据保护条例》[Regulation (EU) 2018/1725,简称 EUDPR][5] 确立了合法性、目的限定、数据最小化和安全保障等一般原则;在此基础上,欧洲数据保护监察机构(European Data Protection Supervisor,EDPS) 提出关于生成式AI 的指引,指引进一步要求:监管机构不得仅凭供应商关于“匿名化”或“合成数据”的声明判断其合规性,而应核验控制措施、风险缓释机制以及各参与方在供应链中的角色定位,并建立处理活动记录,必要时开展数据保护影响评估[6]。该指引同时将模型反演、提示注入、越狱攻击和知识库泄露等生成式AI 特有风险纳入持续监测与红队测试范围;EMA 关于大语言模型的原则文件亦要求严格限定应用场景、数据边界与访问权限[7]。
相比较, 美国的路径则采用技术隔离与用途限制。美国行政管理与预算局(Office of Management and Budget,OMB)备忘录要求联邦机构在使用AI 时同步建立治理框架;FDA内部部署生成式工具时,则明确其运行于高安全级别的政府云环境,并禁止使用监管对象提交的数据训练模型[8]。其制度含义在于,通过切断监管数据进入外部训练闭环的可能性,最直接地保护商业秘密、非公开监管信息和敏感数据,但这一模式的有效性更依赖机构自身的技术能力和内部控制水平。
英国和日本更多依赖既有数据保护法和组织治理安排。前者在英国GDPR 及《数据( 使用与访问) 法案》[Data (Use andAccess) Act] 自动化决策规则基础上强调程序保障[9] ;后者以《个人信息保护法》(Act on the Protection of Personal Information)和日本药品及医疗器械综合机构(Pharmaceuticals and Medical Devices Agency,PMDA)行动计划为基础,突出受控内部环境、组织治理与安全培训[10]。
由此可见,数据安全在药品监管中的核心在于数据进入模型后的边界是否可控、供应链关系是否清晰、处理过程是否留痕可审计以及输出是否仍处于制度可控范围之内。《中华人民共和国数据安全法》《中华人民共和国个人信息保护法》已经对数据处理、合法来源、安全保障和风险处置提出了一般要求,但这些要求仍主要面向一般性数据处理或公众服务场景,尚未细化为药品监管机构内部模型调用、供应链接触、外部训练隔离和结果再利用等高影响场景中的专门要求。未来在药品监管体系中,数据安全的核心已经从“防止信息泄露”转向“控制数据进入模型后的处理边界与决策后果”,其本质是模型接入、供应链管理和监管决策链条完整性的综合治理问题。欧美英日监管数据安全相关制度文件主要内容见表1。

1.2 监管责任与最终责任锚定
当生成式AI 开始参与证据筛选、信号检测和辅助审评时,责任问题便不再只是某一主体是否存在过错,而是开发者、提供者、部署者、使用者以及最终决策者之间如何分配注意义务,以及模型输出如何进入可复核、可审查的行政证据链。换言之,药品监管中的责任问题,真正指向的是“AI 如何被纳入责任可追溯的行政决定结构”。
欧盟通过《欧盟人工智能法案》[Regulation (EU) 2024/1689]与新版《缺陷产品责任指令》[Directive (EU) 2024/2853] 形成了较强的角色化制度安排:前者以风险分级和提供者、部署者等角色义务为中心,要求高风险系统具有人类监督、运行监测和记录留存机制[11] ;后者则将AI 开发者和提供者纳入“制造商”范围,以避免数字技术扩散造成责任真空[12]。这一制度组合的逻辑在于,以法定角色义务将责任前置分配,并通过记录留存和可追溯机制为事后审查、损害救济和行政问责提供接口。
美国尚未形成统一的联邦AI综合立法来直接规定监管机构使用AI 的法律责任, 但OMB 备忘录已将高影响AI 的问责要求落实到具体岗位,并强调人工介入、纠错与救济。FDA 发布的《使用人工智能支持药品和生物制品监管决策的考虑》(Considerations for the Use of Artificial Intelligence to Support Regulatory Decision-Making for Drug and Biological Products)中,也要求对AI 相关输出的采信同步强化记录、复核和解释安排[13],其对监管侧的启示在于:采信或使用AI 相关输出必须同步强化记录、复核与解释安排,以维持行政决定的可审查性。
英国的监管逻辑与之相近,但借用金融界的“沙盒”监管理念,提供了受控的安全测试环境,进而据此提出由行业监管者在具体场景中落实问责要求[14-15]。由此可见,各法域并未赋予AI 独立的行政法地位,而是始终将对外责任锚定于公共机构及其授权人员身上。不同法域的差异不在于“是否问责”,而在于“通过成文立法前置锁定责任”还是“通过治理文件将责任落实到岗位与流程”。我国在《中华人民共和国药品管理法》的框架下,对外发生法律效果的决定同样只能由具有法定职权的监管机构及其工作人员承担[16],AI 可以辅助事实识别、风险提示和技术研判,但不能替代法定主体作出决定。因此,药品监管中真正需要回答的问题,不是“AI 出了错由谁抽象负责”,而是“哪些输出可以进入决定链条、由谁复核、以何种记录方式保证外部可审查”,以及在此基础上如何防止技术参与稀释公共权力的最终责任。欧美英监管责任相关制度文件主要内容见表2。

1.3 数据利用边界与使用权限配置
在药品监管语境中,数据利用边界核心在于监管机构在何种条件下可以调取、整合、处理和再利用健康数据与申报资料, 以及这些数据能否进入模型训练、验证和业务辅助。比较法上的关键问题,不是谁“拥有”数据,而是谁在何种目的、何种环境和何种程序下“ 可以使用”数据。
欧盟在EUDPR 基础上进一步通过欧洲健康数据空间(European Health Data Space,EHDS) 建立“ 许可-目的- 受控环境”的数据二次利用框架,将监管活动以及算法训练、测试和评估纳入允许目的,但要求在安全受控环境中访问数据,并原则上禁止下载原始个人数据[17]。
美国通过《21 世纪治愈法案》(21st Century Cures Act)强化互操作和反信息阻断,扩展可用数据池,但用途边界和责任配置往往仍需在机构治理和具体项目中继续细化[18]。
日本通过《医疗领域研究开发用匿名医疗数据法》(Acton Anonymized Medical Data That Are Meant to Contribute to Research and Development in the Medical Field)和《个人信息保护法》,将来源合法性、主体认证和匿名化加工前置化,以降低数据使用过程中的不确定性[19]。
由此可见,在药品监管场景中,数据利用边界的实质问题在于依法取得的数据能否进一步用于模型训练、模型验证、知识库构建或跨场景调用,以及在这一过程中如何通过用途限定、受控环境和可审计记录维持其合法性基础。我国现行《中华人民共和国数据安全法》《中华人民共和国个人信息保护法》,已经形成“具有合法依据、目的明确、限于必要、来源可审计”的基本要求,为监管数据进入模型提供了原则基础;但现行制度尚未明确,依法取得的申报资料、临床试验数据、药物警戒数据和内部监管资料,能否进一步用于模型训练、模型验证、知识库构建或跨场景再利用,以及相应的程序控制和受控环境要求。也正因此,数据利用边界问题本质上是监管机构使用权限、程序控制和责任配置的公法问题。欧美日监管数据利用边界相关制度文件主要内容见表3。

1.4 隐私保护与程序透明
生成式AI 在药品监管中的隐私风险,已经超出了传统的数据泄露范畴。模型可能在交互中吸收个人信息并在输出中再现,可能通过关联分析形成新的可识别信息,也可能因自动化处理路径不透明而削弱相对人的知情、申辩和救济机会。由此,隐私问题在AI 监管中的意义,不再仅是防止信息外流,更涉及个体权利在自动化决策背景下是否仍能获得实质保障。
欧盟在这一问题上的制度安排最具代表性。EUDPR 不仅强调合法处理与数据主体权利,还严格限制在缺乏实质人类监督时对个人作出具有法律效果或重大类似影响的自动化决定[5]。EDPS 关于生成式AI 的指南进一步强调,应分别识别训练、部署和交互阶段的合法性基础,并核验所部署模型是否存在以不合法方式处理个人数据而开发的风险[6] ;EMA 亦强调在AI开发早期纳入“ 隐私内嵌” 设计和影响评估[20]。美国主要通过《健康保险可携性和责任法案》(Health Insurance Portability and Accountability Act,HIPAA) 及其商业伙伴制度规制受保护健康信息,随后通过《加强电子受保护健康信息网络安全的HIPAA安全规则》(HIPAA Security Rule to Strengthen the Cybersecurity of Electronic Protected Health Information)(草案),拟进一步强化多因素认证、审计日志和完整性验证等网络安全措施托底个人敏感信息的隐私保护[21]。英国强调隐私内嵌、数据最小化和程序保障[22],日本则通过个人信息、假名化信息和匿名加工信息的分层治理,对不同数据形态适用差异化规则[23-24]。对我国而言,《中华人民共和国个人信息保护法》已经确立了自动化决策的透明、公平和说明义务,行政法上的程序公开原则也为AI 介入监管过程中的可解释性和可复核性提供了基础;但现有规则仍相对具有原则性,尚未细化和明确当模型输出实质影响监管判断时,应如何兼顾个人信息保护、程序透明与监管效率。
因此,隐私保护在AI 监管中的真正难点,不在于将旧有规则机械套用于新技术,而在于回应“模型推断能力增强”与“程序可见性下降”同时发生所带来的增量风险。这也解释了为什么各法域都越来越强调人类监督、说明义务、争议机制与救济渠道:在高影响监管场景中,隐私保护已经与程序正当性紧密相连。欧美英日监管隐私保护相关制度文件主要内容见表4。

1.5 知识产权及商业秘密
在药品监管中,知识产权及商业秘密问题并非一般意义上的数据使用权限问题,而是受保护资料进入模型后的限制问题。监管机构即使因法定职责合法接触企业申报资料、技术文件、数据库内容和审评材料,也不当然意味着其可以被无差别用于模型训练、知识库构建或跨场景再利用。欧盟通过《商业秘密指令》[Directive (EU) 2016/943] 保护未公开技术信息和商业信息免受非法获取、使用和披露[25],《思考文件:人工智能在药品生命周期中的应用》(Reflection Paper on the Use of Artificial Intelligence in the Medicinal Product Lifecycle)中也提及AI 处理监管文件时,需关注受保护资料的输入、输出和扩散风险。2025 年FDA 发布的《使用人工智能支持药品和生物制品监管决策的考虑》中表明,AI 输出仅能在具体场景下经可信度评估后用于支持监管决策, 而不能据此推定其可脱离原始资料限制自由流转[13]。日本通过《反不正当竞争法》和《著作权法》维持对技术信息和作品内容的独立保护。我国在知识产权方面已有《中华人民共和国著作权法》《中华人民共和国反不正当竞争法》等一般规则作为支撑, 这意味着企业申报资料、技术文件和数据库内容并非只要技术上可接入模型就可以被合法训练和再利用。现行制度尚未明确监管资料进入模型训练、知识库构建和输出再利用时的授权边界、保密义务和责任配置。
由此,在药品监管AI 应用中,真正需要防止的不是抽象讨论AI 是否享有权利,而在于防止企业申报资料、敏感技术信息和受保护内容在模型训练、系统调用和结果输出过程中被不当吸收、再加工和再扩散。也正因此,知识产权及商业秘密规则在这一领域承担的主要功能,是为监管资料和模型输出设置合法使用边界,从而维护企业合法权益并支撑监管活动的合规性与可归责性。
2 我国药品监管中AI 应用的制度现状与缺口
我国现行法律对于药品监管中AI 的应用并非完全空白。《中华人民共和国数据安全法》《中华人民共和国个人信息保护法》为监管数据处理、敏感个人信息保护和自动化处理提供了一般规则基础;《中华人民共和国药品管理法》确立了药品监管的基本框架和保障用药安全的基本目标;《药品注册管理办法》则在程序层面为AI 嵌入审评审批流程提供了接口。问题不在于“完全无法可依”,而在于现有法律规范主要停留于一般性数据治理和传统行政程序层面,尚未转化为面向监管机构使用AI 的专门制度安排。
总体来看,我国现有制度已经能够为药品监管中的AI 应用提供基本法律支撑,但这种支撑仍以一般法和原则性规则为主,尚未形成针对高影响监管场景的专门治理框架。与前文所归纳的数据安全、监管责任、数据利用边界、隐私保护和知识产权及商业秘密5 类问题相对应,可以发现当前制度缺口主要体现在3 个方面:第一,缺乏面向监管机构使用AI 的专门程序规则,尚未明确不同场景下的适用边界以及哪些事项不得单独依赖模型输出;第二,缺乏AI 输出进入监管证据链的标准,尚未明确模型结果在何种条件下可以被采信,以及应如何复核、解释和留痕;第三,透明度与可信度尚未被固化为一组“可证明材料”与量化指标体系,如模型文档完备率、日志留存周期、复核比例、漂移监测频次、外部审计覆盖率、异议处理时限等。由此可见,我国下一步制度建设的关键,不在于补充零散规则,而在于将现有一般性法律规范进一步细化为适用于药品监管AI 应用的专门治理机制。
3 对我国药品监管制度构建的启示
3.1 比较研究中的制度发现
本节在比较法层面对欧盟、美国、英国、日本等主要法域在药品监管中使用AI 的制度逻辑作集中提炼。就其功能而言,所谓法律问题,主要涉及AI 能否被引入监管活动、基于何种法律依据、在何种用途边界和救济结构下使用、何种输出可以进入监管证据链以及由谁承担最终责任;所谓治理问题,则主要涉及如何将前述要求转化为受控环境、角色分工、日志留痕、版本管理、人类复核、异议处理和退出机制等操作安排。二者并非割裂,而是上位边界与下位控制的关系。进一步看,责任锚定、用途边界、证据采信、权利保护、商业秘密边界等,属于较为稳定的实质问题;部署架构、日志格式、测试频次、审批流程等,则更多属于阶段性的实现问题。
从比较法上看,各法域在药品监管中使用AI 时,虽采取了不同制度路径,但已逐步形成若干可以相互印证的共通趋势。欧盟更倾向于以基本权利保护为起点,通过风险分级、角色义务、影响评估、登记备案、受控环境和数据二次利用许可等制度,将AI 使用纳入“可证明合规”的公法结构;美国则更强调机构治理、公共问责和监管科学,通过治理备忘录、岗位责任、证据记录和特定使用情境下的可信度评估,确保AI 输出只有在可说明、可验证、可复核的条件下,方可进入可审查的行政记录;英国、日本则更多是在既有数据保护、自动化决策和组织治理框架上,叠加人类干预、受控内部环境和安全培训等措施。总体而言,不同法域虽然在规范形式上存在差异,但在高影响监管场景中,均逐步汇聚到责任锚定、边界控制以及透明可审查3项制度重点。
据此,可提炼出3 项制度发现。其一,各法域都未允许AI 分担公共权力的最终对外责任,模型即使参与监管流程,责任仍锚定于监管机构及其授权人员;同时,只有在特定使用情境中可说明、可验证、可复核的输出,才可能进入监管决策链条。这主要对应监管责任问题,也与程序透明要求相衔接。其二,各法域的数据治理重点正从单纯保护数据本身转向控制数据进入模型、在模型中被处理以及在输出环节被再利用的条件,更重视流转边界、用途限制、受控环境和可审计性。这不仅是数据安全,也覆盖数据利用边界及知识产权与商业秘密问题。其三,在高影响监管场景中,透明性与可信度正成为贯穿5 个法律问题的共同抓手。通过模型文档、性能验证、日志留痕、人类复核和异议救济,AI 使用被转化为可证明、可审计、可纠偏的制度安排,这尤其涉及隐私保护及程序透明,并对其余各节形成统摄。
3.2 制度构建路径
对我国而言,药品监管中AI制度构建可沿3 条主线展开。
第一,确立不可削减的共同底线。应明确公共权力的最终责任不得因引入AI 而转移;对审评结论、检查处置和风险预警等高影响事项,不得单独依赖模型输出,必须保留人类复核、申辩与救济机制;凡实质影响监管判断的AI 应用,均应具备用例说明、数据来源和版本记录、模型文档、性能验证、处理日志以及异常退出机制。
第二,坚持场景分层与组合适用。应根据AI 应用对监管判断和相对人权益的影响程度实行差异化治理:高影响场景适用更严格的数据边界控制、用途限制、商业秘密保护、日志留痕和人类复核要求;中低影响场景则可更多依赖事后审计、抽样复核和快速纠偏。
第三,坚持在既有制度基础上叠加治理单元,而非另起炉灶。具体而言,一是明确AI 使用场景、法律依据、用途边界和责任岗位;二是完善模型文档、性能验证、版本管理和证据采信规则;三是将受控环境、训练隔离、商业秘密保护和输出扩散限制嵌入现有数据安全与保密体系;四是将人类复核、说明义务、异议处理和退出机制嵌入行政程序与内部业务规则。
4 结论与展望
总体来看,尽管各法域在药品监管中使用AI 时,围绕数据安全、责任分配、数据利用边界、隐私保护和知识产权及商业秘密所采取的具体规则与制度路径仍存在差异,但其制度演进已经呈现出较为清晰的共同方向:当AI进入监管决策链条后,必须优先完成责任锚定、程序留痕和受控使用,技术效率方可获得法治上的正当性。换言之,AI 在药品监管中的意义,已不再只是提高信息处理能力或优化监管资源配置,而是正在重塑监管决策的形成机制,并由此对监管合法性、可追溯性与可归责性提出更高要求。
对我国而言,下一步制度建设的关键,是在此前针对具体应用场景研究具体细化规则的基础上,着手围绕数据、程序和责任构建一套可组合、可审计、可纠偏的通用治理框架。当AI 进入药品监管高影响环节时,该框架就可以发挥提升监管能力和风险识别效率的技术优势,同时也不会削弱监管决策的合法性、可复核性与公信力。
不仅如此,从当前国际监管协作与全球规则竞争的大背景出发,可持续跟进ICMRA 围绕《药品研发中良好AI 实践指导原则》议题的后续协调进展,并关注国际人用药品注册技术协调会、药品检查合作计划、国际医疗器械监管论坛等主要监管国际规则制定机制中与AI 实际应用密切相关的讨论,从国际规则制定层面审视AI 在药品监管中的应用问题,避免制度设计局限于国内场景而形成“制度孤岛”,而影响未来与国际标准、监管依赖、检查协同和规则互认机制的对接效果。也正是在这个意义上,药品监管中的AI 治理,不应被理解为单纯的技术管理问题,而应被视为一个需要在既有法治框架内持续调适和制度化回应的治理命题,同时也是与国际规则演进保持协调的制度命题。
第一作者简介
王翔宇,博士,国家药品监督管理局科技和国际合作司,助理研究员。专业方向:法学
通讯作者简介
袁林,博士,中国药品监督管理研究会,主任药师。专业方向:药品监管研究
参考文献:略




编辑:李丹
审核:赵燕宜






