 五度妙笔
五度妙笔 企业透视镜
企业透视镜 API商城
API商城
 数据库
数据库Identification of a somatic H3K23me3 methyltransferase SET-19 in
Abstract
Histone methylation plays essential roles in modulating chromatin organization and gene expression. H3K23 methylation is a conserved histone modification, yet its biological roles and the enzymes responsible for its deposition remain poorly understood. Here, we show that the loss of
set-19
leads to a pronounced reduction in H3K23 methylation in
C. elegans
, as revealed by quantitative mass spectrometry, western blotting, and immunofluorescence staining. In vitro biochemical assays show that recombinant SET-19 proteins purified from
E. coli
directly catalyze H3K23 methylation. Genome-wide chromatin immunoprecipitation assays reveal that H3K23me3 is enriched at heterochromatic regions and that loss of
set-19
alters H3K23me3 levels, accompanied by derepression of gene expression. Genetic analyses indicate that SET-19 is dispensable for both germline and somatic RNAi as well as transgenerational epigenetic inheritance of RNAi. SET-19 is predominantly expressed in somatic cells and specifically mediates H3K23me3 deposition in somatic tissues. The loss of
set-19
causes a developmental delay without affecting fertility. Together, our results identify SET-19 as a somatic H3K23 methyltransferase and link H3K23me3 to gene repression in
C. elegans
.
Introduction
The nucleosome, composed of DNA wrapped around histone proteins (H2A, H2B, H3, and H4), is the fundamental structural unit of chromatin. Post-translational modifications (PTMs), such as methylation, acetylation, and phosphorylation, occur on both histone tails and core regions
1
. Histone modifications function as epigenetic marks that regulate chromatin organization and stability, as well as DNA replication, repair, and transcription.
Chromatin is broadly divided into euchromatin and heterochromatin, which differ in their degrees of compaction and transcriptional activity. Histone PTMs such as acetylation, H3K4me3, and H3K36me3 are typically enriched in transcriptionally active euchromatic regions and are associated with gene activation. In contrast, repressive marks including H3K9me3 and H3K27me3 are preferentially enriched in transcriptionally silent heterochromatic regions. The distinct genomic distributions of these modifications reflect their diverse regulatory functions, and the dynamic regulation of these marks across tissues and developmental stages contributes to cell fate determination, fertility, development, and epigenetic inheritance
2
-
6
.
Histone PTMs are dynamically regulated by specific writers, readers, and erasers, which establish, recognize, and remove these modifications, respectively
1
,
7
. Writers responsible for histone methylation are histone methyltransferases (HMTs), which catalyze the methylation of lysine (K) and arginine (R) residues on histone proteins. Lysine residues can be mono-, di-, or trimethylated by lysine methyltransferases (KMTs), whereas arginine residues can be mono- or dimethylated by arginine methyltransferases (RMTs)
8
. Most KMTs harbor a conserved SET domain that confers catalytic activity and substrate specificity
7
,
9
-
11
.
Histone H3 lysine 23 methylation (H3K23me) is widely conserved across eukaryotic species
12
-
24
. In organisms including
Tetrahymena
,
C. elegans
, and mammals, evidence from immunostaining, chromatin immunoprecipitation (ChIP)-based analyses, and binding studies with HP1 family proteins supports an association of H3K23 methylation with heterochromatic regions
14
,
15
,
17
,
18
,
20
,
21
,
24
-
26
. In
Tetrahymena
, the loss of the H3K23 methyltransferase Ezl3p leads to aberrant targeting of meiosis-induced DNA double-strand breaks to heterochromatin and compromises progeny viability
17
. In
C. elegans
, H3K23me3 shares genomic features with classical repressive marks such as H3K9me3 and H3K27me3 and is also associated with transgenerational epigenetic silencing and accumulation at RNAi target loci
25
-
30
. The loss of the corresponding histone methyltransferases for H3K9me3 and H3K27me3 results in varying degrees of RNAi defects
30
-
33
. Two methyltransferases, SET-32 and SET-21, have been reported to contribute to H3K23 methylation
25
,
26
, but no obvious global reduction in H3K23 methylation has been detected in vivo upon loss of either enzyme. Nevertheless, the enzymatic basis, tissue-specific regulation, and biological functions of H3K23 methylation remain far less well characterized than those of other histone methylation marks. The
C. elegans
genome encodes 38 SET domain-containing KMTs
34
-
45
, many of which remain functionally uncharacterized. Our previous work showed that the loss of
set-19
leads to a significant reduction in H3K23me3 levels in vivo
46
, implicating SET-19 as a candidate H3K23 methyltransferase.
In this work, we investigated the role of SET-19 in H3K23 methylation in
C. elegans
and showed that SET-19 functions as a somatic H3K23 methyltransferase required for H3K23me3 methylation in vitro and in vivo. This work provides insights into how H3K23 methylation is established and supports the idea that distinct methyltransferases may function in different tissues or cell types to promote proper development.
Results
SET-19 is required for H3K23 methylation in
C. elegans
In
C. elegans
, the biological functions of many SET domain-containing methyltransferases remain poorly understood
25
,
26
,
34
-
44
. SET-19 harbors a conserved SET domain (
Fig. 1a
). Our previous work showed that the
set-19(ok1813)
mutant exhibited reduced levels of H3K23me2 and H3K23me3, as detected by western blotting
46
. However, the role of SET-19 in H3K23 methylation required further validation.
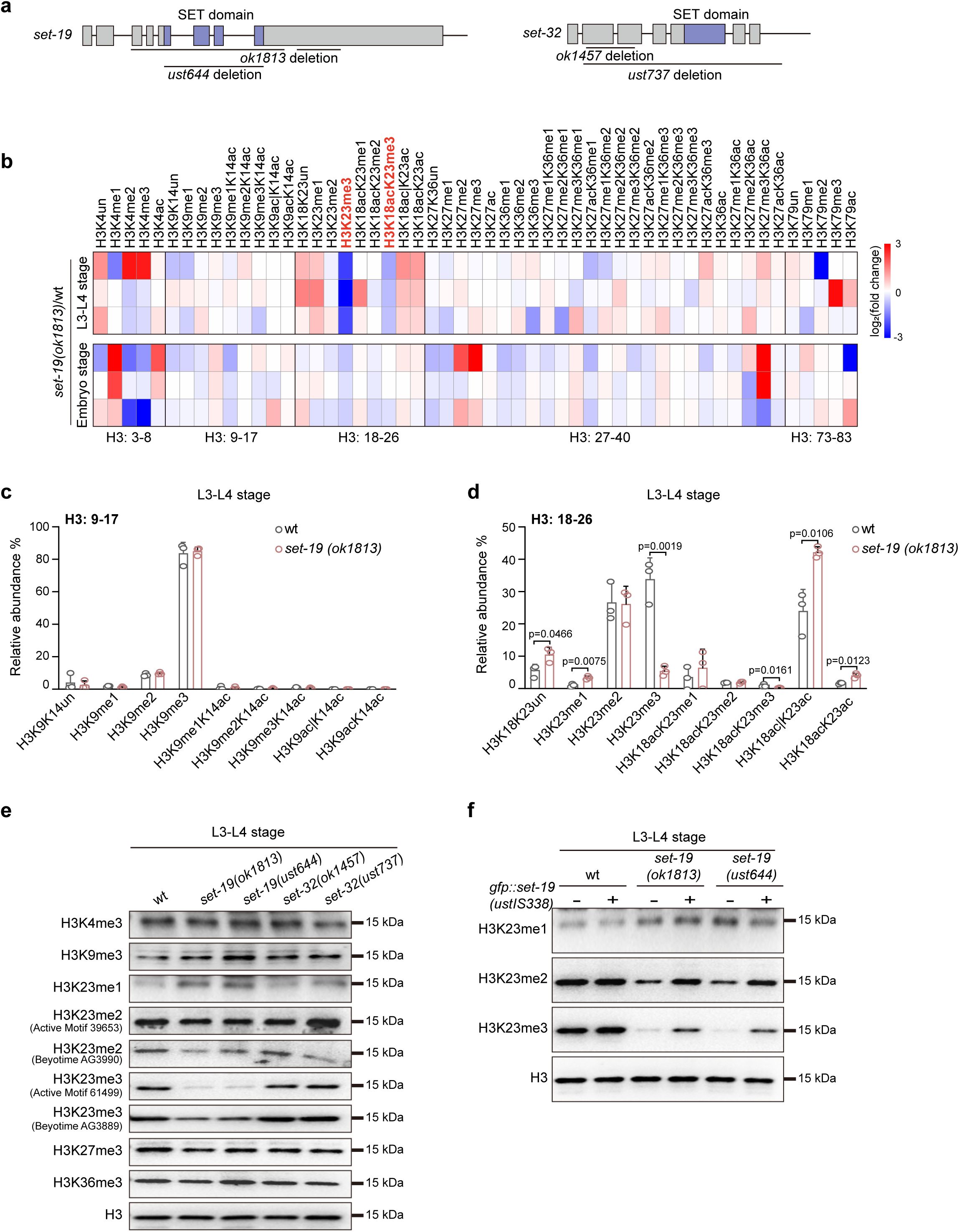
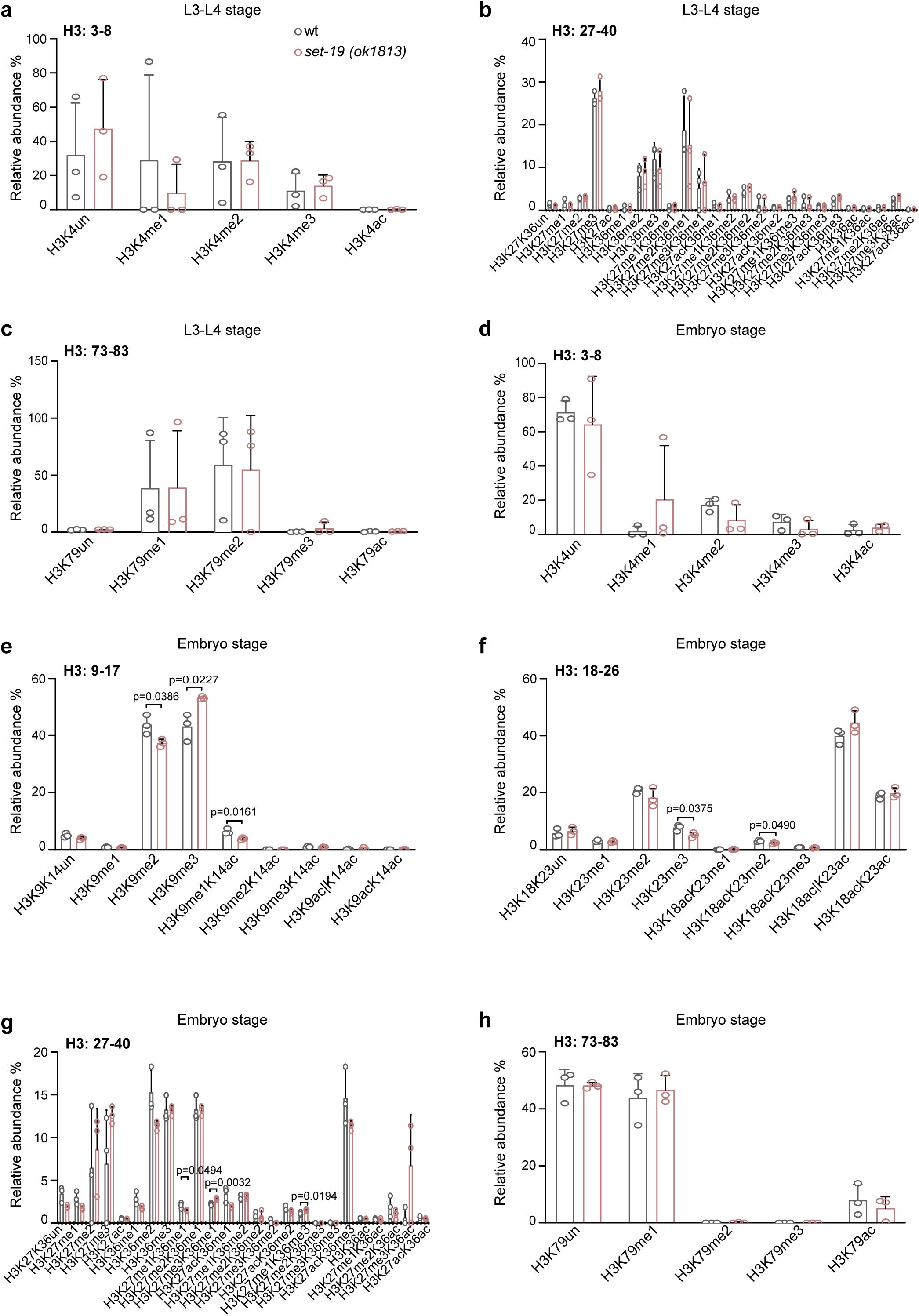
Loss of SET-19 leads to a marked reduction of H3K23me3.
a,
Schematic representation of the
set-19
and
set-32
genes. The
set-19(ok1813)
allele contains two large deletions and likely functions as a null allele. The
set-19(ust644)
allele carries a precise deletion of the SET domain, and the open reading frame is retained. The
set-32(ok1457)
allele contains a deletion that does not disrupt the open reading frame (ORF), whereas
set-32(ust737)
harbors a large deletion and likely functions as a null allele. SET domains are indicated in purple.
b,
Heatmap showing relative abundances of histone H3 post-translational modifications (PTMs) measured by mass spectrometry. Values represent log₂(fold change) in
set-19(ok1813)
mutants relative to wild-type (wt) animals for each biological replicate.
c, d,
Quantification of H3(9–17) and H3(18–26) methylation states in wild-type and
set-19(ok1813)
animals at the indicated developmental stages. Methylation levels are shown as relative abundances, calculated as the area under the curve (AUC) of each modified peptide divided by the sum of all detected forms of that peptide. Differences without indicated p values are not statistically significant (two-tailed t test,
p
< 0.05).
N
= 3 biological replicates.
e,
Western blotting analysis of global H3 methylation levels in L3–L4 stage worms of the indicated genotypes.
f,
Western blotting analysis of H3K23me1/2/3 levels in L3–L4 worms. The
fib-1p::gfp::set-19(ustIS338)
transgene was used for rescue.
To assess the impact of
set-19
loss on histone H3 methylation, we quantified global H3 methylation levels in the
set-19(ok1813)
mutant by mass spectrometry at the L3–L4 larval and embryonic stages. No significant changes were observed in the methylation levels of H3K4, H3K9, H3K27, or H3K36 compared with those in wild-type animals (
Fig. 1b–d
and
Fig. S1
). In contrast, H3K23 methylation was markedly reduced in the
set-19(ok1813)
mutant (
Fig. 1b–d
and
Fig. S1
). Specifically, H3K23me3 levels decreased from approximately 33.8% to 5.4% at the larval stage and from 10% to 8% at the embryonic stage (
Fig. 1d
and
Fig. S1f
). Mass spectrometry revealed no significant change in H3K23me2, whereas H3K23me1 levels were slightly increased at the L3–L4 larval stage (
Fig. 1d
), and a slight reduction of H3K9me2 was detected in embryos (
Fig. S1e
).
To further confirm the role of SET-19, we generated an independent
set-19
allele using CRISPR/Cas9-mediated genome editing. This allele,
set-19(ust644)
, precisely deletes the SET domain without disrupting the open reading frame (ORF) (
Fig. 1a
). Western blotting analysis revealed that both
set-19(ok1813)
and
set-19(ust644)
mutants exhibited a pronounced reduction in H3K23me3 and a detectable decrease in H3K23me2 at the larval stage (
Fig. 1e
and
Fig. S2a
). Two independent antibodies specific for H3K23me2 and H3K23me3, respectively, were used in the western blotting assays. Importantly, reintroduction of a GFP::SET-19 transgene (
fib-1p::gfp::set-19
, ustIS338) into the
set-19
mutant background restored H3K23me2 and H3K23me3 levels (
Fig. 1f
and
Fig. S2b, c
). Consistent with previous reports
25
,
26
, the loss of
set-32
did not lead to a detectable reduction in H3K23 methylation in western blotting assays (
Fig. 1e
and
Fig. S2a
).
Together, these results indicate that SET-19 is required for H3K23 methylation, particularly H3K23me3 in
C. elegans
.
Recombinant SET-19 catalyzes histone H3K23 methylation in vitro
SET-19 contains a central SET domain and a nearby coiled-coil region, with intrinsically disordered regions (IDRs) at both the N- and C-termini (
Fig. 2a
). To test whether SET-19 is a bona fide methyltransferase, we performed in vitro histone methylation assays. Full-length SET-19 and SET-19 domain fragments were fused to GST, expressed in
E. coli
, and purified using glutathione beads. Although full-length SET-19 could not be successfully expressed in
E. coli
and was therefore excluded from subsequent analyses, we obtained purified GST-fused proteins corresponding to the SET domain alone (SET-19 SET; amino acids 37–318) and a fragment containing both the SET and coiled-coil domains (SET-19 SET+CC; amino acids 37–454) (
Fig. 2a
). SET-32 has previously been reported to exhibit H3K23 methyltransferase activity in vitro and was therefore included as a positive control
25
. Given the high evolutionary conservation of histone H3 and the conserved nature of lysine 23 across species (
Fig. S3a
), human histone H3 (Active Motif, #31294) was used as the methylation substrate. Purified GST-SET-19 fusion proteins or full-length GST-SET-32 were incubated with unmodified H3 in the presence of the methyl donor S-adenosylmethionine (SAM). The reaction products were analyzed by western blotting using a panel of antibodies recognizing distinct H3 methylation marks.
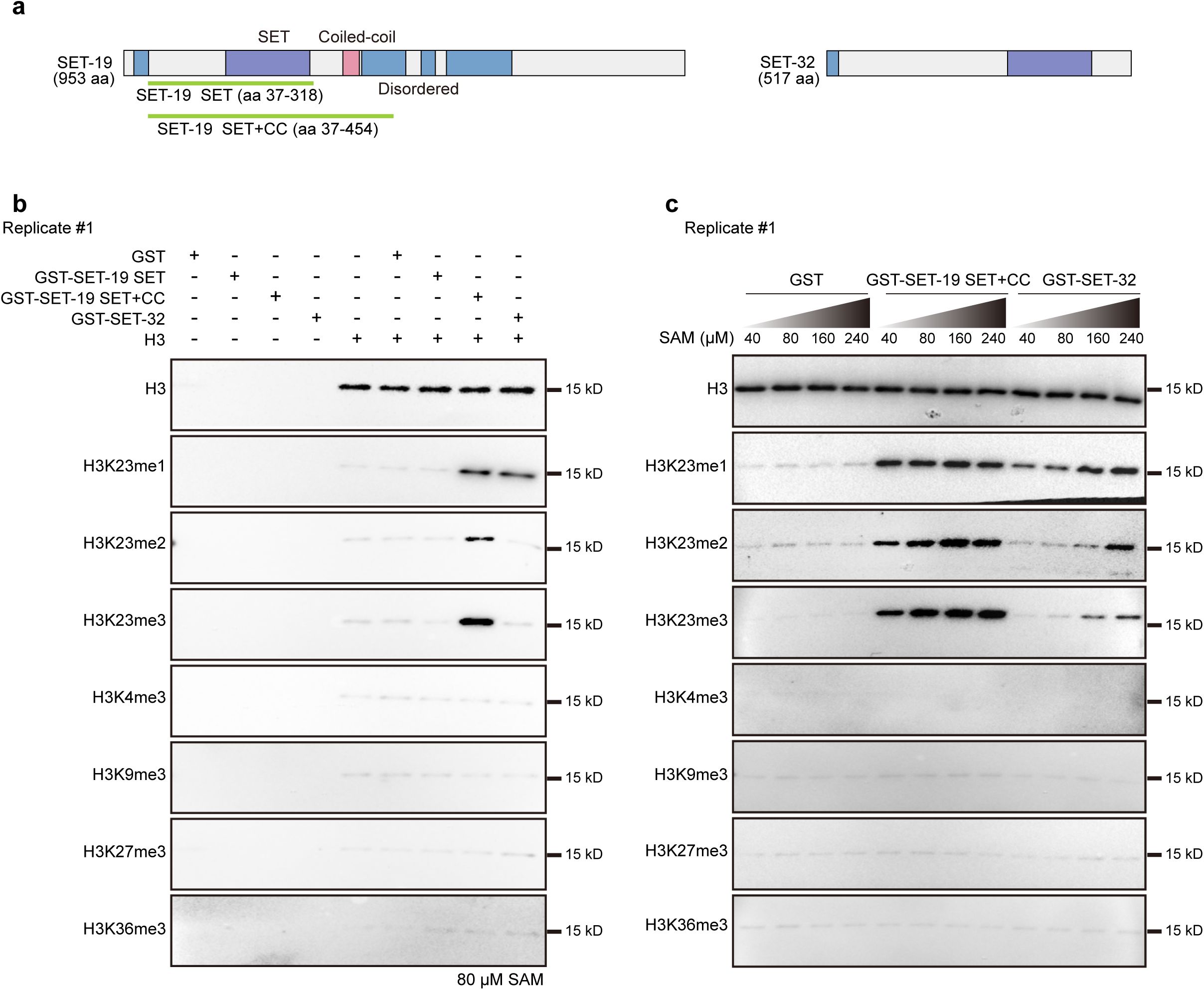
SET-19 specifically catalyzes histone H3K23 methylation in vitro.
a,
Schematic diagram of SET-19 and SET-32 proteins. Purple: SET domain, blue: disordered region, pink: coiled-coil region. Recombinant SET-19 SET or SET+CC fragments are shown in green.
b,
In vitro histone methyltransferase assays using GST-fused SET-19 SET or SET+CC fragments or full-length SET-32 incubated with histone H3 and 80 μM S-adenosylmethionine (SAM). Methylation was detected by western blotting.
c,
In vitro methylation assays performed as in (
b
) with increasing concentrations of SAM.
In the in vitro methylation assays, neither the SET-19 fragments nor full-length SET-32 induced detectable changes in H3K4me3, H3K9me3, H3K27me3, or H3K36me3 levels (
Fig. 2b
and
Fig. S3b
). In contrast, the SET-19 SET+CC fragment, but not the SET domain alone, induced a pronounced increase in H3K23me1, H3K23me2, and H3K23me3 signals (
Fig. 2b
and
Fig. S3b
), indicating that SET-19 specifically methylates H3K23 in vitro and that this activity requires sequences outside the SET domain, which likely helps the folding of the SET domain. SET-32 exhibited modest H3K23me1 methyltransferase activity, but did not appreciably catalyze H3K23me2 or H3K23me3 under the same conditions (
Fig. 2b
and
Fig. S3b
).
SET domain-containing methyltransferases differ not only in substrate specificity but also in their sensitivity to SAM concentration
39
,
47
-
50
. We then examined the enzymatic activities of GST-SET-19 SET+CC and full-length GST-SET-32 across a range of SAM concentrations. Increasing SAM levels enhanced H3K23me1/2/3 signals for both enzymes; however, SET-19 SET+CC consistently exhibited substantially higher catalytic activity than SET-32 (
Fig. 2c
and
Fig. S3c
). No methylation at other lysine residues, including H3K4, H3K9, H3K27, or H3K36, was detected under any of the tested conditions (
Fig. 2c
and
Fig. S3c
). These results indicate that although both SET-19 and SET-32 are capable of catalyzing H3K23 methylation in vitro, SET-19 shows markedly higher enzymatic activity, consistent with the western blotting analyses (
Fig. 1e
).
Notably, although SET-19 catalyzed mono-, di-, and trimethylation of H3K23 in vitro, both mass spectrometry and western blotting analyses revealed a predominant reduction in H3K23me3 upon loss of
set-19
. Together, these in vitro and in vivo data establish SET-19 as an H3K23 methyltransferase in
C. elegans
and indicate that SET-19 plays a prominent role in H3K23 trimethylation.
SET-19 is required for genomic H3K23me3 occupancy and gene repression
Previous studies using ChIP-seq and immunofluorescence staining have shown a strong association between H3K23 methylation (H3K23me2/3) and heterochromatin marks
17
,
18
,
20
,
21
,
24
-
26
. In
C. elegans
, the genome-wide distribution of H3K23me3 closely resembles those of H3K9me3 and H3K27me3 (
Fig. S4a
).
C. elegans
chromosomes are organized into broad domains, with active marks (H3K4me3 and H3K36me3) enriched in central regions and repressive marks (H3K9me1/2/3) enriched in distal arms
34
,
51
-
53
. Consistent with this chromosomal organization, H3K23me3-enriched regions were predominantly localized to chromosome arms (
Fig. S4b
). In addition, regions centered on H3K23me3 peaks show stronger enrichment of H3K9me3 and H3K27me3 than of the active marks H3K4me3 and H3K36me3 (
Fig. 3a
).
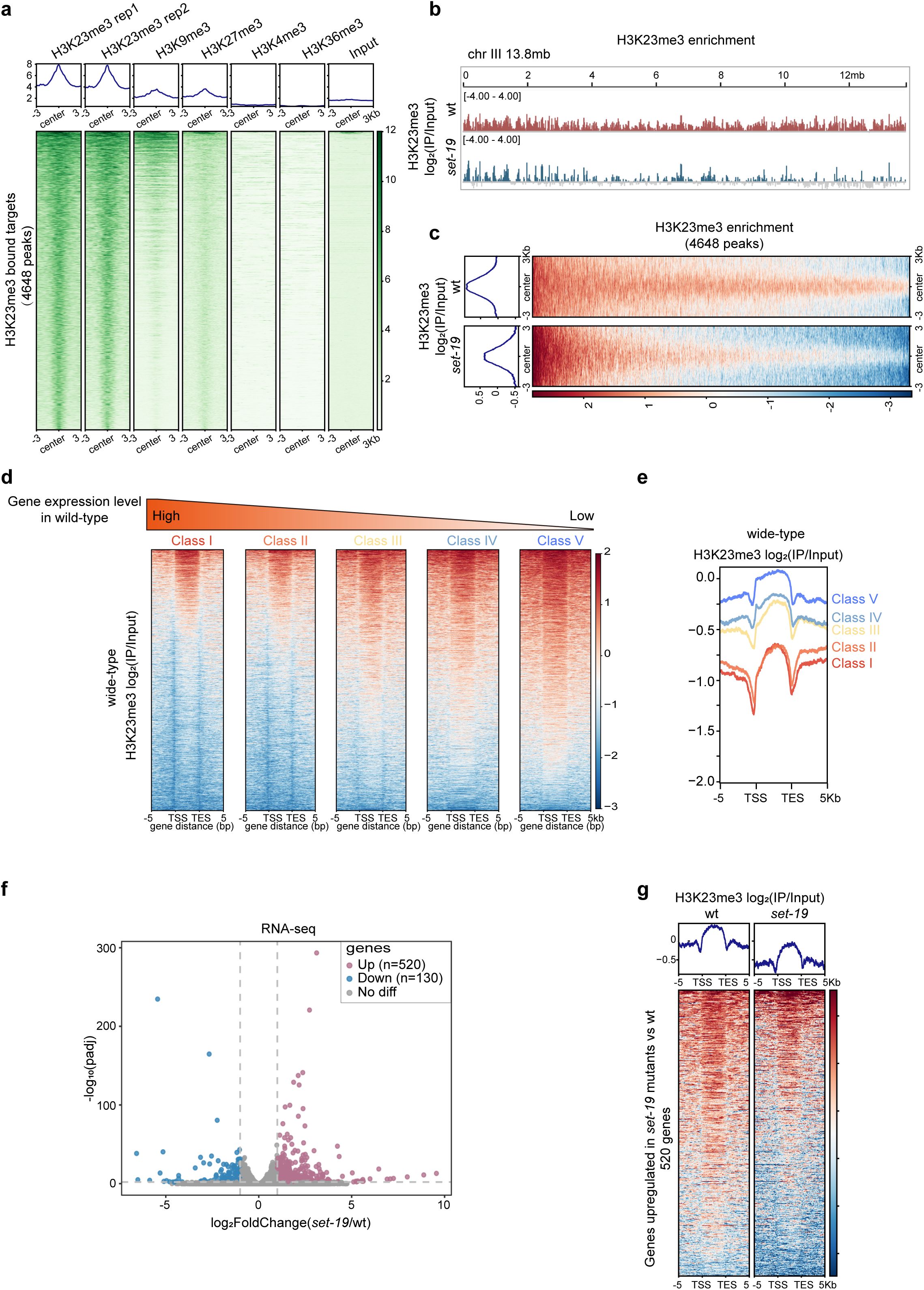
SET-19 is required for genomic H3K23me3 occupancy and transcriptional repression.
a,
Heatmaps showing ChIP-seq signal enrichment of heterochromatic marks (H3K9me3, H3K27me3) and euchromatic marks (H3K4me3, H3K36me3) centered on H3K23me3 peak summits. The average signal within ±3 kb regions is shown.
b,
Genome browser view of H3K23me3 ChIP-seq signals across chromosome III in wild-type and
set-19(ok1813)
animals. Mean log₂ enrichment over input is shown (
N
= 2 biological replicates).
c,
Heatmaps comparing H3K23me3 profiles on H3K23me3 target regions in wild-type and
set-19(ok1813)
worms. Mean log₂ enrichment over input within ±3 kb of peak summits is shown.
d,
Heatmaps of H3K23me3 enrichment across gene bodies in wild-type worms. Genes were grouped into quintiles based on RNA-seq expression levels; genes with zero expression were excluded.
e,
Average H3K23me3 profiles corresponding to the gene groups shown in (
d
).
f,
RNA-seq analysis of
set-19(ok1813)
relative to wild-type worms. Volcano plot showing log
2
FoldChange (
set-19
/wt) versus -log
10
(adjusted p value [padj]), calculated using DESeq2. Significantly upregulated and downregulated genes are highlighted (padj < 0.01; |log
2
FoldChange| > 1).
N
= 3 biological replicates.
g,
H3K23me3 enrichment [log₂(IP/Input)] across gene bodies for genes upregulated in
set-19
mutants. Heatmaps showing a global reduction in the H3K23me3 signal in
set-19(ok1813)
relative to wild-type animals, with average enrichment profiles shown above.
To examine whether SET-19 is required for H3K23me3 occupancy across the genome, we compared H3K23me3 ChIP-seq profiles between wild-type and
set-19
mutant animals. The loss of
set-19
led to a pronounced global reduction in H3K23me3 levels across the genome (
Fig. 3b, c
and
Fig. S4c
). Differential binding analysis using DiffBind
54
revealed that 55.4% of H3K23me3 peaks were significantly reduced in the
set-19
mutant relative to wild-type animals (
Fig. S4d
). Notably, residual H3K23me3 signals remained in the
set-19
mutant (
Fig. 3b, c
and
Fig. S4c, d
), suggesting additional H3K23 methyltransferases besides SET-19 may also contribute to H3K23 methylation.
H3K23me3 has been shown to be required for transcriptional repression of nuclear RNAi-targeted genes
25
,
26
. To investigate the contribution of SET-19-dependent H3K23me3 to gene regulation, we performed mRNA-seq analysis in wild-type N2 and
set-19
mutant animals. Genes were grouped into five categories based on their expression levels in wild-type N2 animals, and then H3K23me3 levels were quantified for each group. H3K23me3 levels showed a strong negative correlation with gene expression, with lowly expressed genes exhibiting higher H3K23me3 occupancy (
Fig. 3d, e
). Transcriptome analysis revealed that 520 genes were significantly upregulated and 130 genes were downregulated in the
set-19
mutant compared with wild-type animals (
Fig. 3f
). Importantly, the upregulated genes displayed reduced H3K23me3 levels in the
set-19
mutant (
Fig. 3g
), indicating that the loss of SET-19-dependent H3K23me3 is associated with transcriptional derepression.
Collectively, these results indicate that SET-19 is required for genomic H3K23me3 occupancy and is linked to transcriptional repression.
SET-19 is dispensable for feeding RNAi targeting both germline and somatic genes
H3K23me3 and the putative H3K23 methyltransferase SET-32 have previously been implicated in transgenerational epigenetic inheritance (TEI)
25
,
26
,
31
,
33
,
55
. We first tested whether SET-19 is required for feeding RNAi targeting several germline and somatic genes, including
pos-1
,
mex-3
,
dpy-11
, and
unc-15
. RNAi targeting
pos-1
or
mex-3
causes embryonic lethality and results in unhatched F1 embryos
56
-
58
. RNAi targeting
dpy-11
or
unc-15
induces dumpy and paralysis phenotypes, respectively
59
-
61
. However, neither
set-19
nor
set-32
mutants displayed detectable defects in these feeding RNAi responses (
Fig. S5a–d
). RDE-1 is an essential Argonaute protein required for feeding RNAi response
62
. As a control,
rde-1
mutants were completely resistant to feeding RNAi. To increase the sensitivity of feeding RNAi assays, we next used an enhanced RNAi background
60
. In
eri-1
mutants, RNAi targeting
dpy-11
resulted in a strongly enhanced dumpy phenotype in the progeny. RNAi targeting
lir-1
induced larval arrest, and targeting
dpy-13
induced a super dumpy phenotype
60
,
63
,
64
; both phenotypes were abolished by
nrde
mutation. However, the
eri-1;set-19
mutant remained fully competent for feeding RNAi against
dpy-11
,
lir-1
, and
dpy-13
(
Fig. S5e–g
).
We then asked whether SET-19 also participates in the transgenerational epigenetic inheritance of RNAi. We employed a germline-expressed
pie-1
p
::h2b::gfp
reporter and performed feeding RNAi against
gfp
for one generation, after which animals were transferred to fresh plates without continued RNAi exposure. As expected, wild-type animals silenced
gfp
expression in the parental generation and maintained robust silencing in their F1 progeny. In
hrde-1
,
set-25
, and
set-32
mutants, the parental generation remained sensitive to feeding RNAi, but silencing inheritance was abolished in the F1 progeny (
Fig. 4a, b
). In contrast,
set-19
mutants retained effective
gfp
silencing in both the parental and F1 generations, comparable to that in control animals (
Fig. 4a, b
). Similarly, we tested whether SET-19 is required for the intergenerational inheritance of RNAi using a soma-expressed
sur-5p::gfp
reporter. Feeding dsRNA targeting
gfp
resulted in efficient silencing of GFP expression in both parental animals and F1 progeny in control animals (
Fig. 4c
). As expected, GFP expression was not silenced in the
rde-1
mutant, consistent with the essential role of RDE-1 in feeding RNAi responses. NRDE-3 is required for the intergenerational inheritance of RNAi targeting somatic genes
60
. In
nrde-3
mutants, the somatic
sur-5p::gfp
was silenced in the parental generation, but this silencing was abolished in the F1 progeny. In
set-19
and
set-32
mutants,
sur-5p::gfp
remained silenced in both parental and progeny generations (
Fig. 4c, d
).
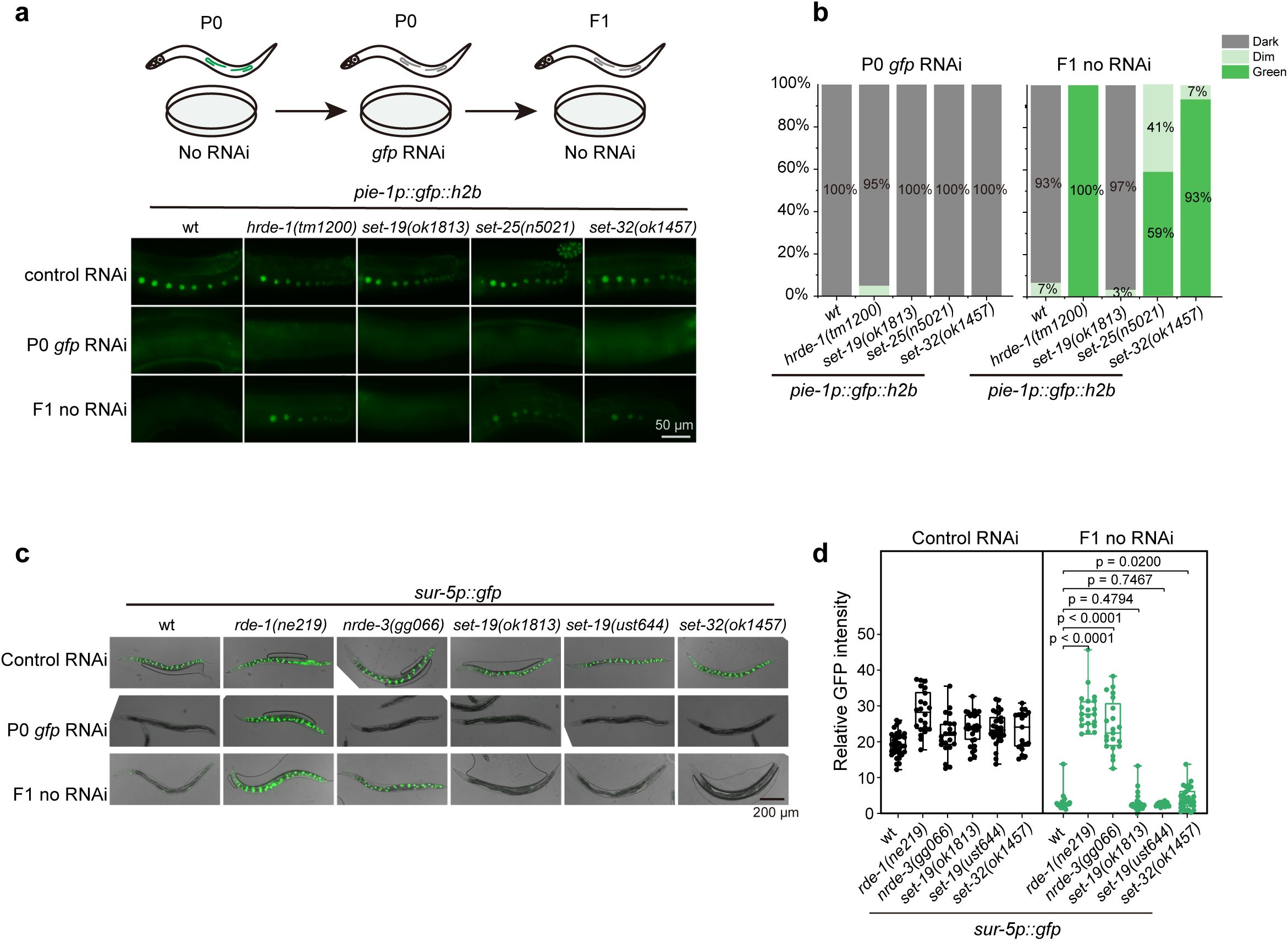
SET-19 is dispensable for feeding RNAi and its inheritance.
a,
Representative fluorescence images of animals expressing
pie-1
p
::gfp::h2b
subjected to
gfp
RNAi or control treatment. F1 progeny were maintained on control bacteria without feeding RNAi.
b,
The percentages of the indicated P0 and F1 animals expressing GFP were quantified. At least 30 animals were scored per genotype and generation.
c,
Representative fluorescence images of animals expressing
sur-5p::gfp
following
gfp
RNAi treatment.
d,
Quantification of relative GFP fluorescence intensity for animals shown in (
c
). Data are presented as the mean ± SD (
N
= 3 biological replicates; n > 18 worms per genotype). Statistical significance was assessed using a two-tailed t test.
Thus, these results suggest that SET-19 is dispensable for RNAi and its inheritance in
C. elegans
.
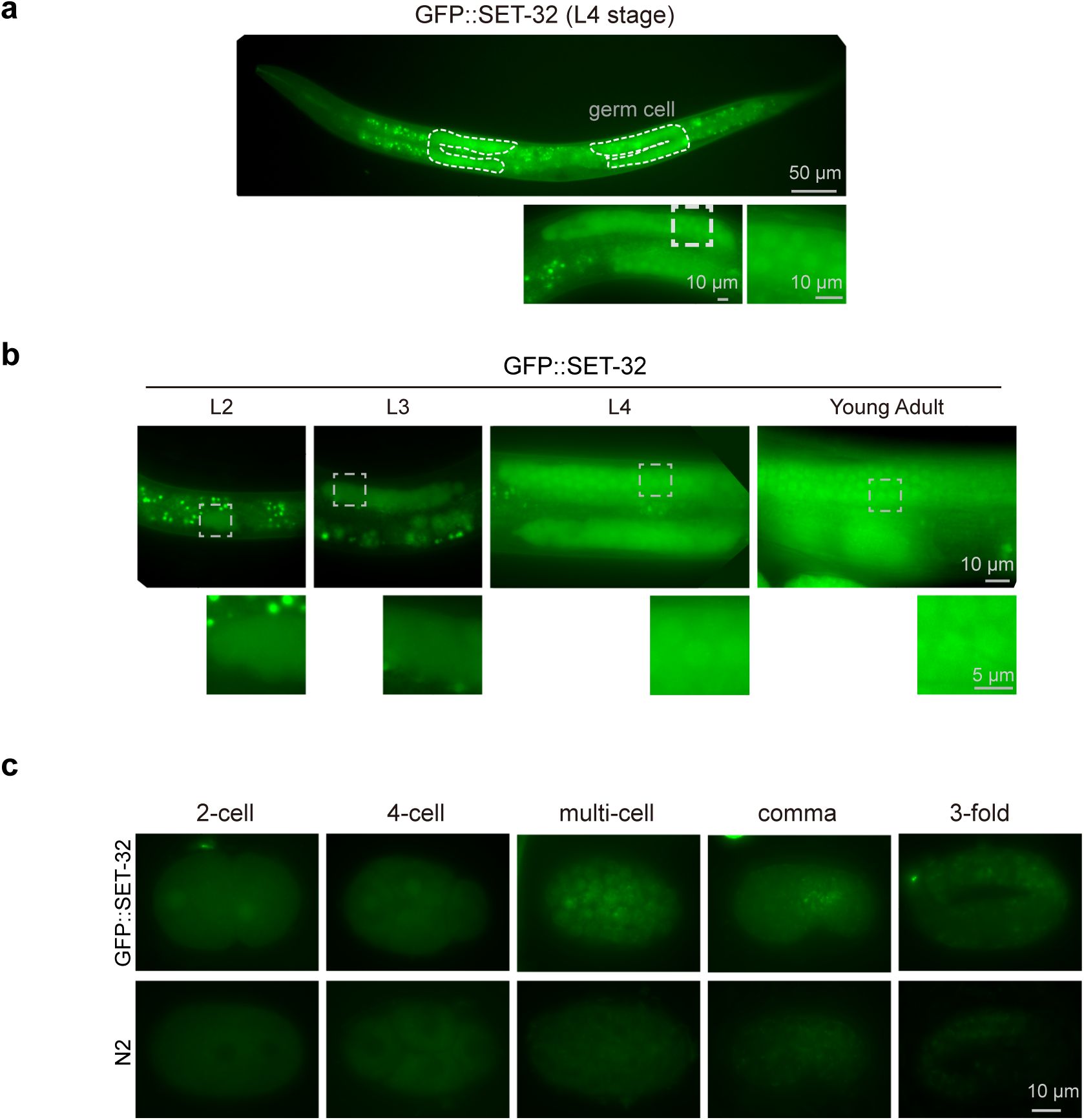
SET-19 is predominantly expressed in somatic cells and required for developmental timing
To explore the physiological roles of SET-19, we examined the development and reproduction of
set-19
mutants, but did not observe significant defects in brood size or hatch rate compared with wild-type animals (
Fig. 5a, b
). However,
set-19
mutants exhibited a modest developmental delay relative to wild-type animals (
Fig. 5c
).
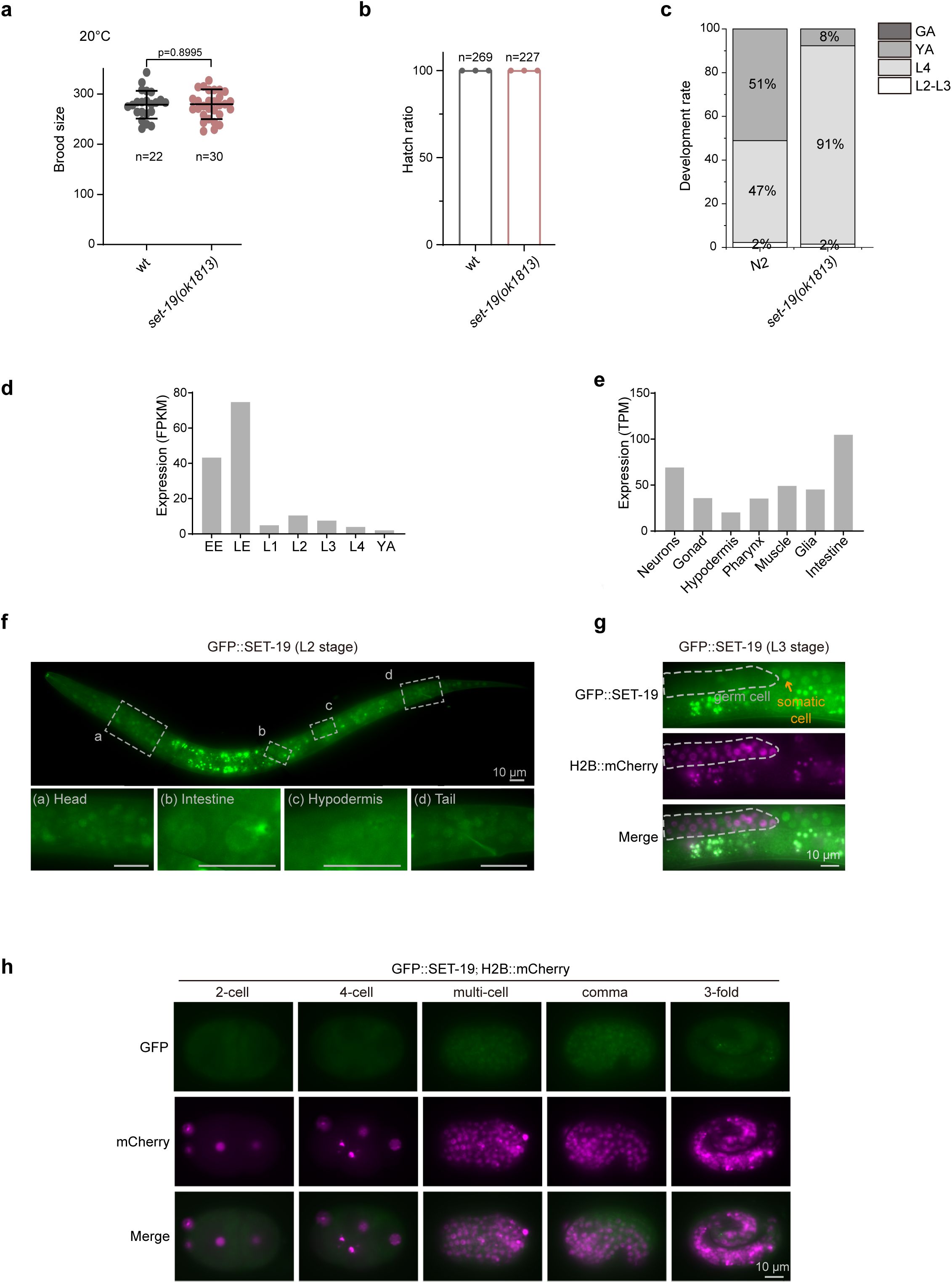
SET-19 is expressed in somatic cells and required for developmental timing.
a,
Brood size of the indicated genotypes maintained at 20°C. Individual L4 animals were transferred to fresh NGM plates, and total progeny were counted.
b,
Hatch rate of embryos laid by synchronized adult hermaphrodites at 20°C. The hatch rate was calculated as the ratio of hatched larvae to total embryos.
c,
Developmental progression of the indicated genotypes at 20°C (n > 50).
d,
The expression levels of
set-19
mRNA at different developmental stages. Data were downloaded from WormBase (version WS297). EE, early embryos; LE, late embryos; YA, young adults.
e,
The expression levels of
set-19
mRNA in different tissues at the L2 stage
65
.
f,
Fluorescence images of L2 animals expressing GFP::SET-19.
g,
Fluorescence images of L3 animals expressing GFP::SET-19 and H2B::mCherry. GFP::SET-19 shows enrichment in somatic cells.
h,
Subcellular localization of GFP::SET-19 and H2B::mCherry in embryos.
Publicly available expression data from WormBase (version WS297) showed that
set-19
mRNA is more highly expressed in embryos than in larvae (
Fig. 5d
). To further examine cell type-specific expression, we analyzed published single-cell RNA-seq data from L2-stage larvae
65
.
set-19
expression was relatively enriched in intestinal cells and neurons (
Fig. 5e
).
To directly assess the expression and subcellular localization of SET-19, we generated an endogenous GFP-tagged SET-19 strain using CRISPR/Cas9-mediated genome editing. GFP::SET-19 was broadly expressed in somatic cells during both embryonic and larval stages and was predominantly localized to the nucleus (
Fig. 5f–h
). During embryogenesis, SET-19 expression was low in early embryos but markedly increased in late embryos (
Fig. 5h
). No detectable GFP::SET-19 signal was observed in germ cells (
Fig. 5g
). In contrast, SET-32 was highly expressed in germ cells, displayed both nuclear and cytoplasmic localization during larval stages, and was weakly expressed in embryos (
Fig. S6a–c
).
These results indicate that SET-19 is predominantly expressed in somatic cells and is largely excluded from the germline.
SET-19 conducts H3K23 trimethylation predominantly in somatic cells
To further examine the role of SET-19 in H3K23me3 in vivo, we performed immunofluorescence staining and assessed H3K23me3 levels across different developmental stages and tissues. In wild-type N2 animals, H3K23me3 signals were readily detected throughout development and were broadly distributed across tissues (
Fig. 6a–d
). Consistent with the somatic expression pattern of SET-19, the loss of
set-19
resulted in a pronounced reduction in H3K23me3 in somatic cells (
Fig. 6a
). However, no obvious reduction of H3K23me3 was observed in embryos (
Fig. 6b
). In young adult worms, the reduction in H3K23me3 was most prominent in intestinal cells (
Fig. 6c
), a tissue in which
set-19
expression is relatively enriched (
Fig. 5e
). Notably, H3K23me3 levels in germ cells remained largely unchanged in
set-19
mutants (
Fig. 6d
), consistent with the absence of detectable
set-19
expression in the germline (
Fig. 5g
).
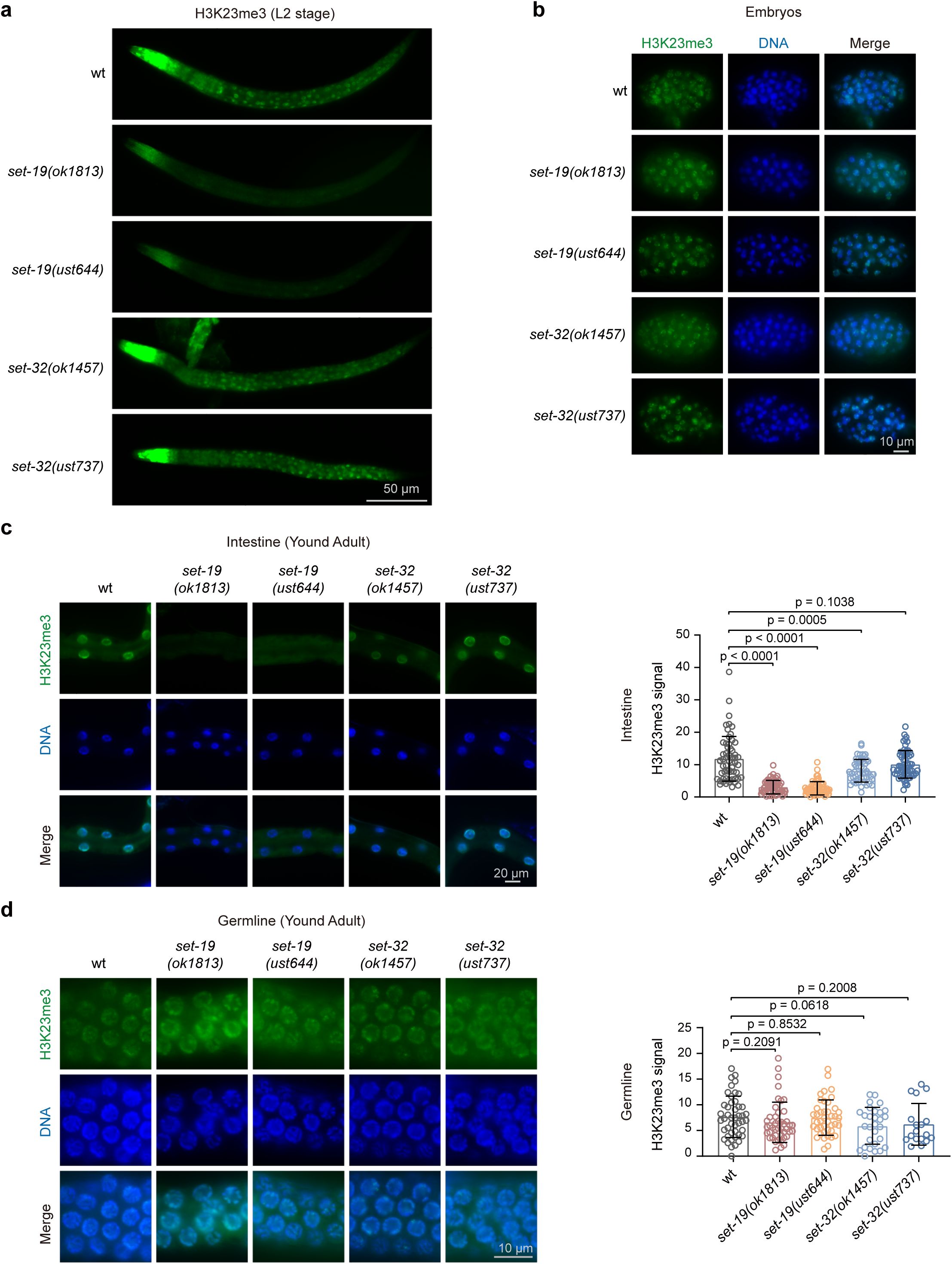
SET-19 mediates H3K23me3 predominantly in somatic cells.
a,
Representative immunofluorescence images of H3K23me3 staining in L2-stage worms.
b,
Representative H3K23me3 immunostaining in embryos.
c,
Representative images (left) and quantification of H3K23me3 immunostaining in the intestinal nuclei of young adult worms (right). H3K23me3 levels were calculated as nuclear fluorescence intensity after background subtraction, with three nuclei averaged per worm. H3K23me3 (green); DAPI (blue). Signal quantification was performed using ImageJ. A two-tailed t test was performed to determine statistical significance. For intestinal nuclei: wild type,
n =
60 worms,
N =
3 biological replicates;
set-19(ok1813)
,
n =
60 worms,
N =
3 biological replicates;
set-19(ust644)
,
n =
60 worms,
N =
3 biological replicates;
set-32(ok1457)
,
n =
51 worms,
N =
2 biological replicates;
set-32(ust737)
,
n =
60 worms,
N =
3 biological replicates.
d,
Representative images (left) and quantification of H3K23me3 immunostaining in the germline nuclei of young adult worms (right). H3K23me3 levels were calculated as nuclear fluorescence intensity after background subtraction, with three nuclei averaged per worm. H3K23me3 (green); DAPI (blue). Signal quantification was performed using ImageJ. A two-tailed t test was performed to determine statistical significance. For germline nuclei: wild type,
n =
46 worms,
N =
3 biological replicates;
set-19(ok1813)
,
n =
44 worms,
N =
3 biological replicates;
set-19(ust644)
,
n =
42 worms,
N =
2 biological replicates;
set-32(ok1457)
,
n =
29 worms,
N =
2 biological replicates;
set-32(ust737)
,
n =
18 worms,
N =
2 biological replicates.
For comparison, we analyzed H3K23me3 levels in
set-32
mutants. The loss of
set-32
resulted in no, if any, detectable reduction in H3K23me3 across tissues or developmental stages (
Fig. 6a–d
), suggesting that SET-32 is unlikely to act as the major H3K23me3 methyltransferase in
C. elegans
.
Together, these results support a major role for SET-19 in establishing H3K23me3 in somatic cells.
Discussion
H3K23me3 is a histone modification presented across multiple species, yet its mechanisms of deposition and biological functions have remained poorly defined. In this study, we identified SET-19 as an H3K23me3 methyltransferase through a combination of biochemical and in vivo approaches. We show that SET-19-dependent H3K23me3 is enriched at heterochromatin-associated regions and is associated with transcriptional repression. SET-19-dependent deposition of H3K23me3 occurs predominantly in somatic tissues in
C. elegans
. Collectively, these findings extend our understanding of how H3K23 methylation is established and suggest that its deposition is regulated in a tissue-specific manner.
Histone lysine methylation is generally catalyzed by SET domain-containing methyltransferases. The
C. elegans
genome encodes at least 38 SET domain-containing lysine methyltransferases, yet only a subset of them have been functionally characterized
35
-
44
,
66
-
69
. To date, efforts to identify the enzymes responsible for H3K23 methylation have been relatively limited. Candidate-based analyses assessed H3K23me2 by immunostaining in primordial germ cells and the adult germline, but failed to identify a bona fide KMT for germline H3K23me2
18
. SET-32 was then shown to methylate H3K23 in vitro and to be required for nuclear RNAi-induced H3K23me3 in vivo
25
,
26
. Subsequently, the closely related methyltransferase SET-21 was also shown to possess H3K23 methyltransferase activity in vitro, predominantly generating H3K23me1 and H3K23me2. SET-32 and SET-21 are more strongly associated with the germline
26
. Yet single loss of
set-32
or
set-21
did not cause a noticeable reduction in overall H3K23 methylation. Here, we show that SET-19 functions as a new H3K23me3 methyltransferase that acts primarily in somatic tissues. Based on our findings together with previous reports, we therefore propose a working model in which SET-19 predominantly mediates H3K23me3 deposition in somatic cells, whereas germline H3K23 methylation may rely more heavily on SET-32 and SET-21 (
Fig. 7
).
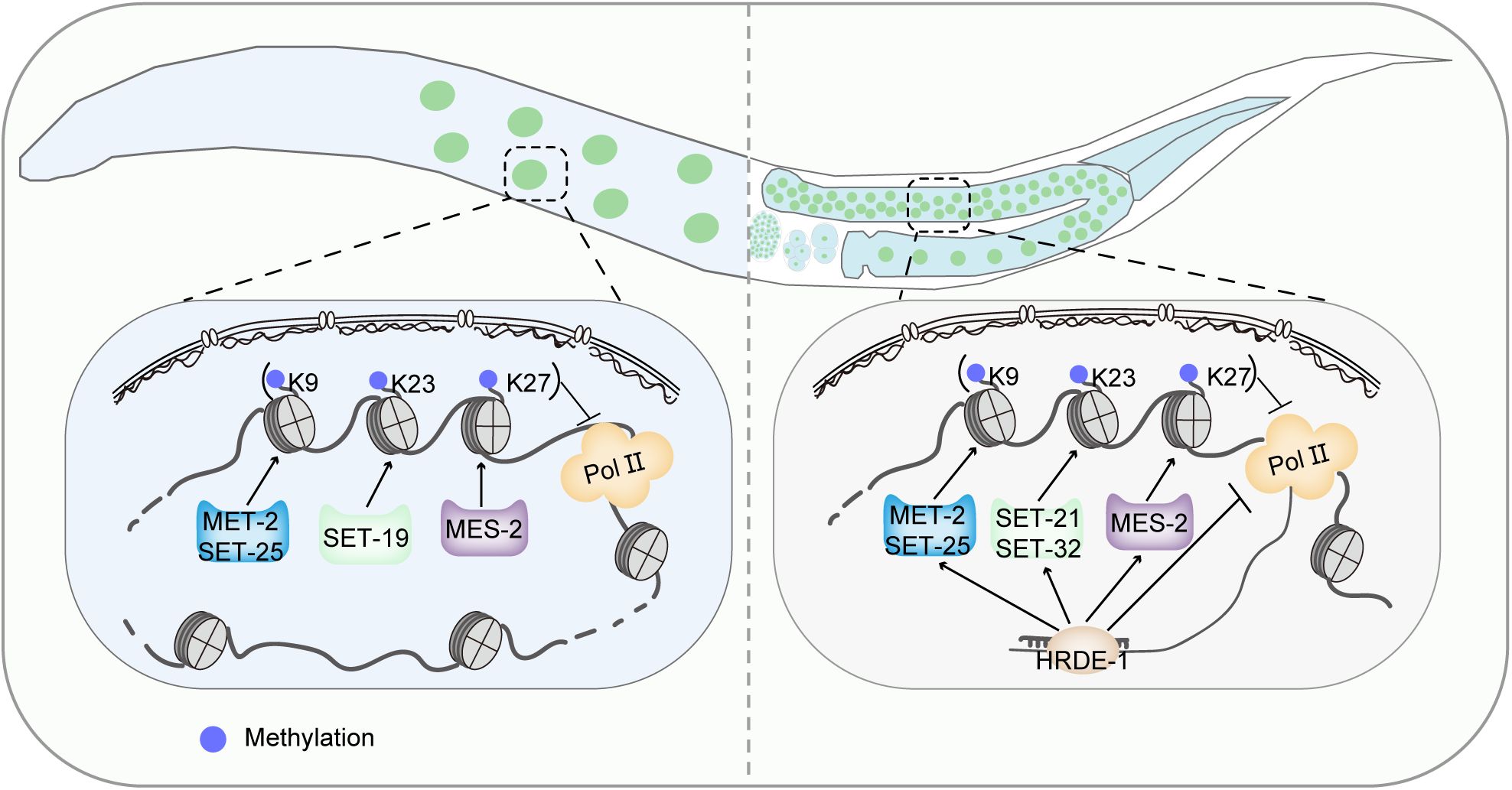
A working model for somatic and germline H3K23 methylation in
C. elegans
.
(Left) In somatic cells, SET-19 acts as the major H3K23 methyltransferase. SET-19-catalyzed H3K23me3 coexists with H3K9me3 and H3K27me3 in heterochromatic regions and represses transcription. (Right) In the germ line, SET-32 and SET-21 participate in the maintenance of H3K23me3 downstream of the HRDE-1-mediated RNAi pathway and contribute to the transgenerational inheritance of RNAi.
Classical heterochromatin marks, such as H3K9 and H3K27 methylation, play essential roles in transcriptional repression, perinuclear anchoring of chromatin, genome stability, and RNAi-mediated gene silencing
27
-
30
,
70
-
72
. Previous studies linked H3K23me3, particularly SET-32-associated H3K23 methylation, to nuclear RNAi and transgenerational epigenetic inheritance
25
,
26
,
29
,
31
,
33
,
73
. Immunofluorescence staining shows that H3K23me3 forms discrete foci within the nucleus, resembling the nuclear distribution patterns reported for H3K9 and H3K27 methylation
74
. In addition, the heterochromatin protein HPL-1, a chromodomain-containing reader, has been shown to recognize both H3K9 and H3K23 methylation
18
. Our data show that H3K23me3 is enriched at heterochromatin-associated regions and is linked to transcriptional repression. These observations suggest that H3K23me3 is an additional heterochromatin-associated repressive mark. Nevertheless, whether H3K23 methylation contributes to perinuclear chromatin organization remains an intriguing question. Future studies using perinuclear tethering assays could be used to assess the contribution of H3K23 methylation to nuclear positioning
74
.
H3K23 methylation appears to be functionally distinct from other heterochromatin marks. The major H3K9 and H3K27 methyltransferases in
C. elegans
, MET-2/SET-25 and MES-2, respectively, act broadly across tissues and developmental stages
42
,
75
. In contrast, SET-19 contributes to H3K23me3 predominantly in somatic cells, pointing to distinct modes of enzymatic regulation among repressive histone modifications. This tissue-specific pattern of deposition further raises the possibility that different H3K23 methyltransferases may make distinct functional contributions. Although SET-32 has been linked to nuclear RNAi and transgenerational epigenetic inheritance in the germline
25
,
26
,
29
,
31
,
33
,
73
,
set-19
mutants do not exhibit noticeable defects in feeding RNAi targeting either germline or somatic genes. These results indicate that SET-19 is not an indispensable requirement for the execution of feeding RNAi and its inheritance. However, whether SET-19 contributes to RNAi-induced H3K23me3 deposition in somatic cells requires further investigation.
While our findings support a predominantly somatic role for SET-19, the functions of H3K23 methylation in both somatic and germline cells remain to be fully defined. The unresolved questions include whether multiple H3K23 methyltransferases act redundantly or in parallel in the germline. It also remains unclear how SET-19-dependent and germline-enriched H3K23 methyltransferases are coordinated across tissues, and to what extent H3K23me3 overlaps functionally with other heterochromatin machineries. Systematic biochemical and genetic analyses of SET domain proteins in
C. elegans
will help define the full complement of histone methylation activities and clarify how chromatin-based regulation is directed to distinct developmental stages and cell types.
Materials and Methods
Strains
The Bristol strain N2 was used as the standard wild-type strain. All strains were grown at 20°C unless otherwise specified. The strains used in this study are listed in
Table S1
.
Construction of plasmids and transgenic strains
For in situ expression of GFP::SET-19, the coding sequence of 3×FLAG::GFP, fused to a linker sequence (
GGAGGTGGAGGTGGAGCT
), was inserted immediately downstream of the start codon of
set-19
using the CRISPR/Cas9 system. The 3×FLAG::GFP fragment was PCR-amplified from YY178 genomic DNA. Homologous left and right arms (1.5 kb each) were amplified from N2 genomic DNA, and the backbone fragment was amplified from plasmid pCFJ151. All fragments were assembled into the repair plasmid using the ClonExpress MultiS One Step Cloning Kit (C113-02, Vazyme) via Gibson assembly. The injection mixture contained pDD162 (50 ng/µl), the repair plasmid (50 ng/µl), pCFJ90 (5 ng/µl), and two or three sgRNA plasmids targeting sequences near the N-terminus of the
set-19
gene (20 ng/µl per sgRNA plasmid). The mixture was injected into adult animals. Three to four days after injection, F1 worms expressing pharyngeal GFP were isolated using a Leica M165 FC fluorescence stereomicroscope. Progeny from these F1 worms were subsequently screened by PCR and validated by Sanger sequencing. The sequences of primers used for construction of the in situ transgenic strain are listed in
Table S2
.
Construction of mutant strains via CRISPR/Cas9 technology
To generate sgRNA expression vectors, the 20 bp
unc-119
sgRNA guide sequence within the
pU6::unc-119
sgRNA(F+E) vector was replaced with distinct target-specific sgRNA guide sequences. A plasmid mixture containing 30 ng/µl of each of the three or four sgRNA expression vectors, 50 ng/µl of pDD162 plasmid, and 5 ng/µl of pSG259 was co-injected into wild-type N2 nematodes. Worms carrying the desired gene deletions were screened via PCR following the method described previously
76
. All sgRNA sequences used in this study are listed in
Table S3
.
Isolation of histones and mass spectrometry
Synchronized L3–L4 larvae and embryos (obtained by bleaching gravid adults) were washed three times with 1× M9 buffer. All samples were flash-frozen in liquid nitrogen and pulverized into a fine powder using a ball mill (60 Hz). The frozen powder was resuspended in Nuclear Extraction (NE) Buffer (100 mM HEPES, pH 7.5; 50 mM NaCl; 1% Triton X-100; 0.1% sodium deoxycholate; 1 mM EDTA; 10% glycerol; plus protease inhibitors). The resulting suspension was centrifuged at low speed to remove worm debris. To remove soluble proteins, the clarified supernatant was further centrifuged at high speed (16,000 × g for 10 min), and the resulting supernatant was discarded. The insoluble pellet was resuspended in NE buffer. Histone isolation was performed using a high-salt extraction strategy
77
. First, the insoluble pellet was further lysed in salt-free buffer to disrupt nuclear envelopes; histones were subsequently liberated from chromatin complexes via extraction with 2.5 M NaCl buffer. Histones were separated by SDS-PAGE, and the Coomassie blue-stained band containing histone H3 was cut from the gel. The gel slices underwent two rounds of propionylation with propionic anhydride to derivatize free and monomethylated lysine residues. Afterward, the samples were digested with trypsin, followed by an additional propionylation step to modify newly exposed N-terminal amines
39
,
78
.
The eluted peptides were analyzed on an Easy-nLC 1000 system (Thermo Fisher) coupled to a Q Exactive mass spectrometer (Thermo Fisher) by LC-MS/MS. The acquired RAW data were analyzed using EpiProfile 2.1_Celegans, an updated version of EpiProfile 2.0
79
. For each histone modified peptide, the relative abundance (%RA) was calculated by dividing the area under the curve (AUC) of each modified peptide by the sum of the areas corresponding to all the observed forms of that peptide.
Western blotting
Synchronized L3–L4 larvae and embryos obtained by bleaching gravid adults were washed three times with 1× M9 buffer. Samples were stored at -80°C until use. The samples were suspended in 2× SDS loading buffer and heated in a metal bath at 95°C for 5–10 minutes. The suspensions were then centrifuged at 16,000 × g, and the supernatants were collected. Proteins were resolved by SDS-PAGE on gradient gels (15% separation gel, 5% spacer gel) and transferred to a Hybond-ECL membrane. After washing with 1× Tris-buffered saline with Tween-20 (TBST) buffer and blocking with 5% milk-TBST, the membrane was incubated overnight at 4°C with antibodies. The membrane was washed three times for 10 minutes each with 1× TBST and then incubated with secondary antibodies at room temperature for 2 hours. The membrane was washed thrice for 10 minutes with 1× TBST and then visualized. The following antibodies were used for western blotting: anti-H3 (Abcam, ab1791), 1:8000; anti-H3K4me3 (Abcam, ab8580), 1:800; anti-H3K9me3 (Millipore, 07-523), 1:3000; anti-H3K27me3 (Millipore, 07-449), 1:8000; anti-H3K36me3 (Abcam, ab9050), 1:1000; anti-H3K23me1 (Active Motif, 39388), 1:6000; anti-H3K23me2 (Active Motif, 39653), 1:2000; anti-H3K23me2 (Beyotime, AG3990), 1:2000; anti-H3K23me3 (Active Motif, 61499), 1:3000; and anti-H3K23me3 (Beyotime, AG3889), 1:1000. Secondary antibodies: HRP-labeled goat anti-rabbit IgG (H +L) (Abcam, ab205718), 1:15000.
Recombinant protein expression and purification
The DNA segments encoding the SET domain of SET-19 (SET-19 SET; aa 37–318), the SET and coiled-coil domains of SET-19 (SET-19 SET+CC; aa 37–454) and the full-length protein of SET-32 were amplified from a cDNA library of wild-type strain N2 using PCR. Subsequently, the SET-19 SET, SET-19 SET+CC and SET-32 constructs were cloned and inserted into the pGEX-4T-1 plasmid and expressed in
E. coli
BL21-GOLD (DE3) cells (Novagen). The recombinant proteins were affinity-purified via GST tag binding to glutathione beads (Smart-Lifesciences, SA008100) according to the manufacturer’s instructions. The eluted protein was dialyzed against a buffer containing 20 mM Tris-HCl pH 7.5, 5% glycerol, and 200 mM NaCl and subsequently concentrated. The concentrated protein was then subjected to high-speed centrifugation; after centrifugation, the clarified protein solution was supplemented with glycerol and stored at -80°C.
In vitro histone methylation assay
GST-fused proteins (0.25 µM) were incubated with 1 µM histone H3 (Active Motif, 31294) and 80 µM S-adenosylmethionine (SAM) (NEB, B9003S) in a 20 µl reaction mixture containing methyltransferase activity buffer (20 mM Tris-HCl, pH 8.0; 50 mM NaCl; 20 mM KCl; 10 mM MgCl₂; 5% glycerol; 0.02% Triton X-100; 0.1 mg/ml BSA; and 1 mM dithiothreitol [DTT]) for 3 h at 20°C. Reactions were terminated by the addition of SDS loading buffer, followed by boiling for 15 min. Proteins were resolved on a 15% SDS-PAGE gel and analyzed by western blotting.
Imaging
For imaging larval stages, animals were immobilized in 0.1 M sodium azide and mounted on 1.5% agarose pads. For imaging embryos and germ cells, gravid adults were dissected on a coverslip in 2 μl of 0.4× M9 buffer containing 0.1 M sodium azide and then mounted on freshly prepared 1.1% agarose pads. Imaging was performed using a Leica THUNDER Imaging System equipped with a K5 sCMOS camera and HC PL FLUOTAR objectives (100×/1.40–0.70 oil, 40×/0.80, and 20×/0.80). Images were acquired using Leica Application Suite X software (v63).
Immunofluorescence staining and quantification
Embryos were freeze-cracked using liquid nitrogen, then fixed in methanol for 30 seconds and post-fixed in 1% paraformaldehyde (PFA) for 2 minutes. After three washes in PBS containing 0.25% Triton X-100 (PBS-T), samples were permeabilized in PBS with 1% Triton X-100 for 20 minutes. Subsequently, the samples were blocked in PBS containing 0.5% BSA before overnight incubation with the primary antibody against H3K23me3 (Active Motif, 61499) at 4°C. Following three washes with PBS-T, samples were incubated with secondary antibodies (Alexa Fluor 488-conjugated anti-rabbit) for 2 hours at room temperature, before three final PBS-T washes and DNA staining with Hoechst 33342. For germline and intestine staining, gravid adult worms were dissected in M9 buffer, fixed in methanol for 1 minute at -20°C and in 2% PFA for 5 minutes. For L2 worm staining, worms were freeze-cracked using liquid nitrogen, then fixed in 1% paraformaldehyde (PFA) for 20 minutes. Fixed animals were permeabilized by sequential thiol-based reduction and oxidation steps. Samples were incubated in PBS containing 10% β-mercaptoethanol for 15 minutes, treated with PBS containing 10 mM DTT for 15 minutes, and then incubated in PBS containing 0.3% H₂O₂ for 15 minutes. The animals were subsequently fixed in methanol for 30 seconds and treated with the permeabilization solution (PBS containing 1% Triton X-100) for 20 minutes. Images were taken on a Leica THUNDER Imaging System with identical gain settings within each experimental set. Quantification was performed using ImageJ by measuring the mean gray value within the nuclear region and that of an adjacent background area. For each worm or tissue, fluorescence intensity was calculated as the average nuclear signal from three nuclei, subtracting the average background signal from three corresponding adjacent background regions. At least two independent experiments were performed.
Brood Size
L4-stage hermaphrodites were singled onto plates and transferred daily as adults until embryo production ceased, and the progeny numbers were scored.
Hatch ratio
Synchronized adult hermaphrodites were transferred to NGM plates to lay eggs, which were subsequently removed. The numbers of embryos and hatched larvae were scored, and the hatch ratio was calculated as the number of hatched larvae divided by the total number of embryos.
Development rate
Synchronized adult hermaphrodites were transferred to NGM plates to lay eggs, which were subsequently removed. The growth of the progeny was monitored every 24 hours at 20°C.
RNAi
RNAi experiments were performed at 20°C by placing synchronized embryos on RNAi feeding plates as previously described
80
. HT115 bacteria expressing the empty vector L4440 (a gift from A. Fire) were used as controls. Bacterial clones expressing target-specific double-stranded RNAs (dsRNAs) were obtained from the Ahringer RNAi library
81
and their identities were verified by Sanger sequencing.
Total RNA isolation
Synchronized L4-stage worms were washed three times with 1× M9 to remove bacterial residues. For RNA extraction, 400 µl of TRIzol Reagent (Ambion, 15596026) was added to a 100 µl worm aliquot, followed by 7–8 cycles of freezing in liquid nitrogen and thawing in a 42°C water bath. Afterward, 100 µl of DNA/RNA Extraction Reagent (Solarbio LIFE SCIENCES, P1014) was added to the samples, which were then centrifuged at 16000 × g for 15 minutes at 4°C. The collected supernatant was further treated with 400 µl isopropanol and 400 µl pre-chilled 75% ethanol, and subjected to DNase I digestion (Thermo Fisher Scientific, EN0521). Finally, RNA was eluted into 20 µl of nuclease-free water and used for mRNA library preparation.
mRNA-seq analysis
The Illumina-generated raw reads were first filtered to remove adapters, low-quality reads, and contaminants, yielding high-quality clean reads. The clean reads were aligned to the reference genome of WBcel235 via HISAT2 (v2.1.0)
82
. For quantification, featureCounts (v1.6.0)
83
was used to calculate gene-level read counts. Library size factors were calculated using DESeq2 (v1.46.0)
84
with its standard median-of-ratios method based on genome-wide gene expression counts. Genes with an adjusted
P
value (padj) < 0.01 and an absolute log2FoldChange > 1 were considered significantly differentially expressed. Three biological replicates were analyzed per condition. All statistical analyses and plots were generated using custom R scripts.
Chromatin immunoprecipitation (ChIP)
ChIP experiments were performed as previously described
30
. For L3–L4 worms, frozen animals were first ground to a fine powder in liquid nitrogen
85
. Then, the samples were crosslinked in 2% formaldehyde for 10 minutes at room temperature with gentle rotation. Crosslinking was quenched by adding 0.125 M glycine (final concentration) for 5 minutes at room temperature. Samples were sonicated for 13 cycles (30 s on and 30 s off per cycle) at high output with a Bioruptor Plus (Diagenode), with the sample tubes kept in an ice-water bath throughout the process. The lysates were precleared and immunoprecipitated with a rabbit anti-H3K23me3 antibody (Active Motif, 61499) overnight at 4°C. Chromatin/antibody complexes were recovered with Dynabeads
TM
Protein A (Invitrogen, 10002D) followed by extensive sequential washes with 150 mM, 500 mM, and 1 M NaCl, respectively. Crosslinks were reversed overnight at 65°C. The recovered DNA was treated with RNase (Roche) for 30 minutes at 65°C, and all DNA samples were purified using a DNA purification kit (TianGen, #DP204).
ChIP-Seq
The DNA samples from the ChIP experiments were subjected to quality control (QC) prior to sequencing and then sequenced at high depth at Novogene Bioinformatics Technology Co., Ltd. (Beijing, China). Briefly, the ChIP DNA was processed for library construction following the standard protocol: the DNA fragments were combined with an End Repair/A-tailing mix and incubated in a single reaction to complete end repair and 3’-end adenylation. The resulting A-tailed DNA fragments were then incubated with sequencing adapters in ligation mix to achieve adapter ligation. The adapter-ligated DNA was subjected to size selection and several rounds of PCR amplification to obtain the final DNA library. The size distribution of the library fragments was analyzed on the Agilent 5400 system (Agilent, USA; with matching Agilent assay reagents). The library was quantified by qPCR to a final concentration of 1.5 nM. The qualified libraries were pooled according to their effective concentration and required data volume and then further amplified on cBot to generate clusters on the flow cell and sequenced with a paired-end 150 bp (PE150) method on an Illumina platform.
Published ChIP-seq datasets of histone modifications in
C. elegans
were downloaded from the NCBI GEO or ENCODE databases. The datasets used in this study are listed in
Table S4
.
ChIP-seq data analysis
ChIP-seq data analysis was performed as previously described
86
. ChIP-seq reads were aligned to the WBcel235 assembly of the
C. elegans
genome using Bowtie2 (v)
87
by Ben Langmead with the default settings. The SAMtools (v0.1.19)
88
“view” utility was used to convert the alignments to BAM format, and the “sort” utility was used to sort the alignment files. ChIP-seq peaks were called using MACS2 (v2.1.1)
89
with subcommand. For the H3K4me3 and H3K36me3 datasets, which display narrow enrichment patterns, peaks were identified using the parameters “-g ce -B -f BAM -q 0.01”. For the H3K27me3, H3K9me3, and H3K23me3 datasets, which show broad histone modification domains, peaks were identified using the parameters “-g ce -B -f BAMPE --broad --broad-cutoff 0.01”. Deeptools subcommand bamCoverage (v3.5.0) was used to produce bigWig tracks for data visualization with defined parameters (--binSize 20 --normalizeUsing BPM --smoothLength 60 --extendReads 150) from bam files. The Integrative Genomics Viewer genome browser
90
was applied to visualize signals and peaks. The genomic regions were defined as chromosome arms and centers based on previous works
51
. For visualization of peak locations, we used the R package ggplot2 (v4.0.0).
For heatmap analysis, the called peaks of H3K23me3 rep1 were used. Deeptools subcommand computeMatrix (v3.4.3) was used to calculate the score matrix for the heatmap with defined parameters (reference-point --referencePoint center -b 3000 -a 3000 --skipZeros). The heatmap was plotted with Deeptools subcommand plotHeatmap (v3.5.0).
ChIPseqSpikeInFree normalization for H3K23me3
H3K23me3 ChIP-seq experiments were performed with two replicates each in both wild-type (wt) and
set-19
mutant backgrounds. To compare the H3K23me3 signals between the two backgrounds in the absence of spike-in, ChIPseqSpikeInFree (v1.2.4)
91
was used to calculate the scaling factor (SF) for every sample (SF
i
). The effective ChIP-seq library size for sample i was calculated as N
i
∗ SF
i
, where N
i
is the original library size. The effective library size was then used to normalize the read count from sample i during the downstream differential analysis and heatmap visualization. For the two replicates of wt and
set-19
mutants, pairwise comparisons were performed, and the resulting SF values were averaged, yielding SF_wt = 1 and SF_set-19 = 2.115. The “-scale” parameter for bedtools genomecov was calculated from the effective library size using the formula: Scale parameter = 15000000/(Ni × SFi), where 15000000 is the preset reference read count. Normalized BEDGraph files were generated by setting the “-scale” parameter to the value from the above formula. The genome was then partitioned into fixed 20 bp windows, and the normalized signal was mapped into these windows using a bedtools map with weighting by the actual overlap length. The log₂(IP/Input) enrichment value for each window was subsequently calculated after adding a pseudocount of 1 to both IP and Input signals to avoid log₂(0) errors. Finally, the sorted BEDGraph file was converted into BigWig format for visualization. For the differential analysis of H3K23me3 peaks, we used the R package DiffBind (v3.16.0)
54
and integrated the sample-specific SF into the total read count normalization via the underlying DESeq2 (v1.46.0)
84
framework.
Statistics
Bar graphs with error bars are presented with mean and standard deviation (SD). All experiments were conducted with independent
C. elegans
animals for the indicated N times. Statistical analysis was performed with a two-tailed Student’s t test.
Figure supplements
Mass spectrometry analysis of global H3 methylation in
set-19(ok1813)
mutants.
Quantification of H3 methylation levels in wild-type and
set-19(ok1813)
mutants by quantitative mass spectrometry. H3 methylation levels were quantified as relative abundances, calculated by dividing the area under the curve (AUC) of each modified peptide by the sum of the AUCs of all observed forms of that peptide and multiplying by 100. Differences without indicated p values are not statistically significant (two-tailed t test,
p
< 0.05).
N =
3 biological replicates.
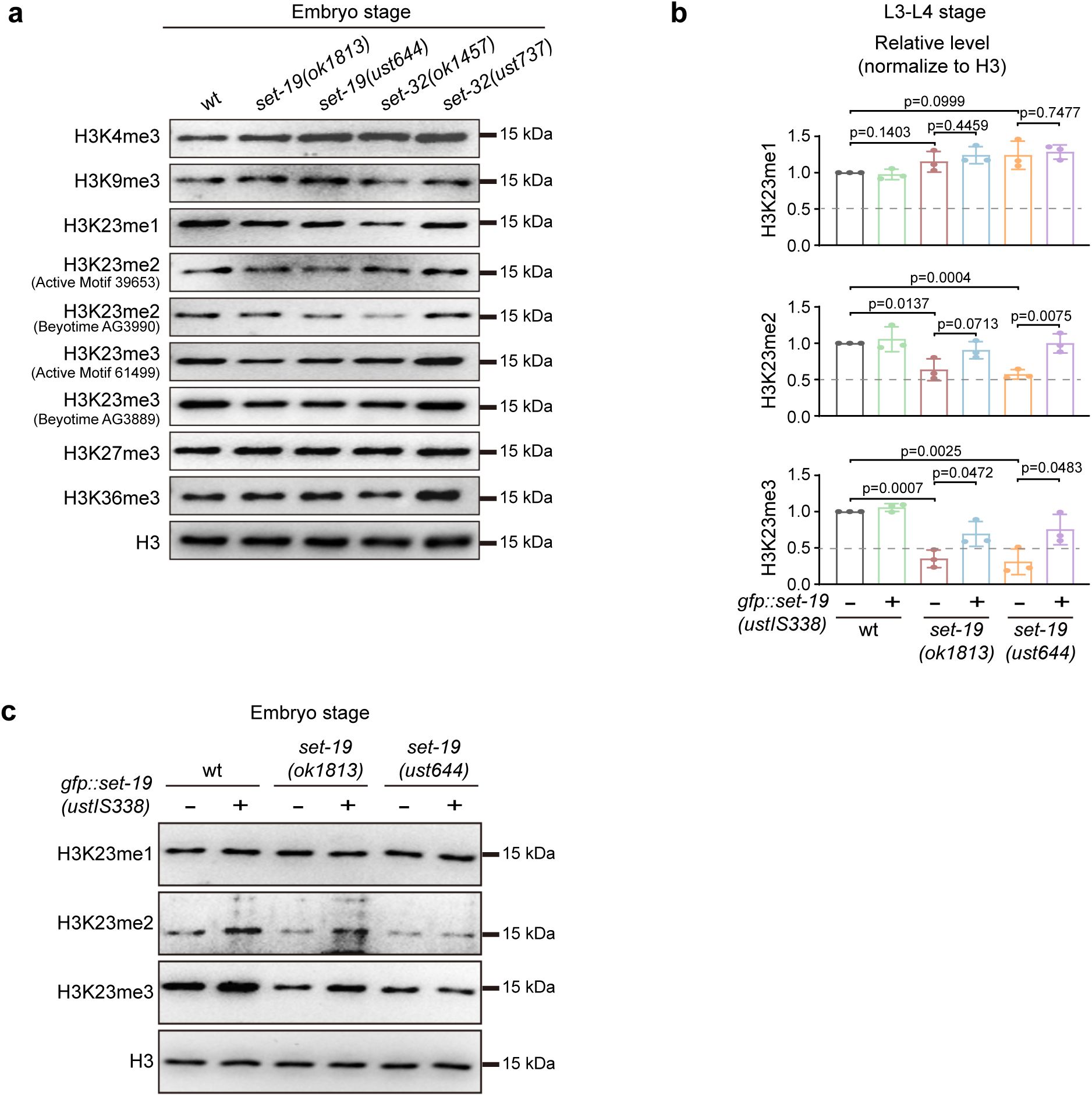
The loss of
set-19
reduces H3K23 methylation levels.
a,
Western blotting analysis of global H3 methylation levels in embryos of the indicated genotypes.
b,
Quantification of H3K23me1/2/3 levels in L3–L4 worms, related to
Fig. 1f
.
N =
3 biological replicates; two-tailed t test.
c,
Western blotting analysis of H3K23me1/2/3 levels in embryos. The
fib-1p::gfp::set-19(ustIS338)
transgene was used for rescue.
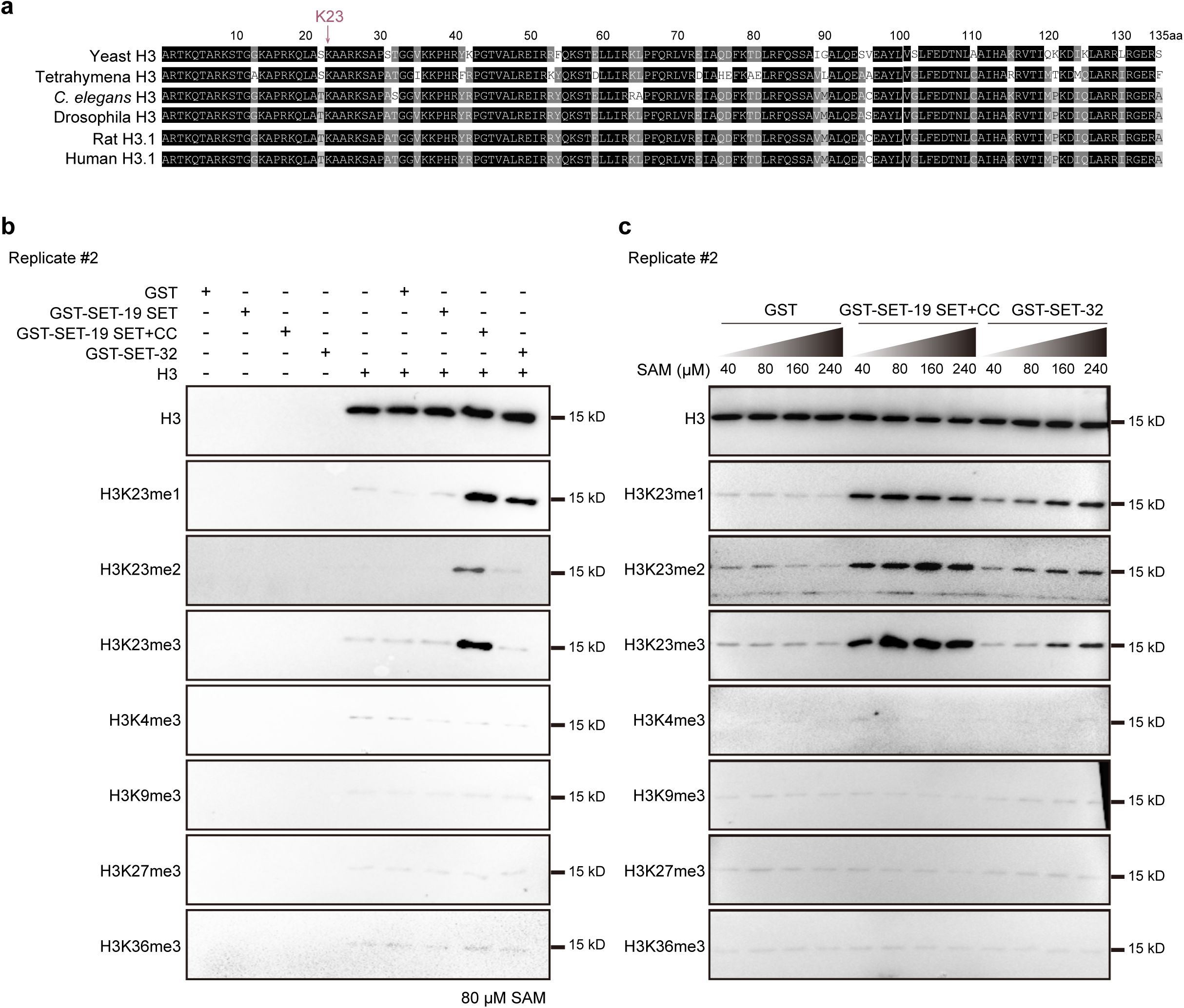
SET-19 specifically catalyzes histone H3K23 methylation in vitro.
a,
Multiple sequence alignment of histone H3 proteins across species, highlighting the conserved lysine 23 residue.
b, c,
Independent replicates of in vitro methylation assays corresponding to
Fig. 2b
and
Fig. 2c
, respectively.
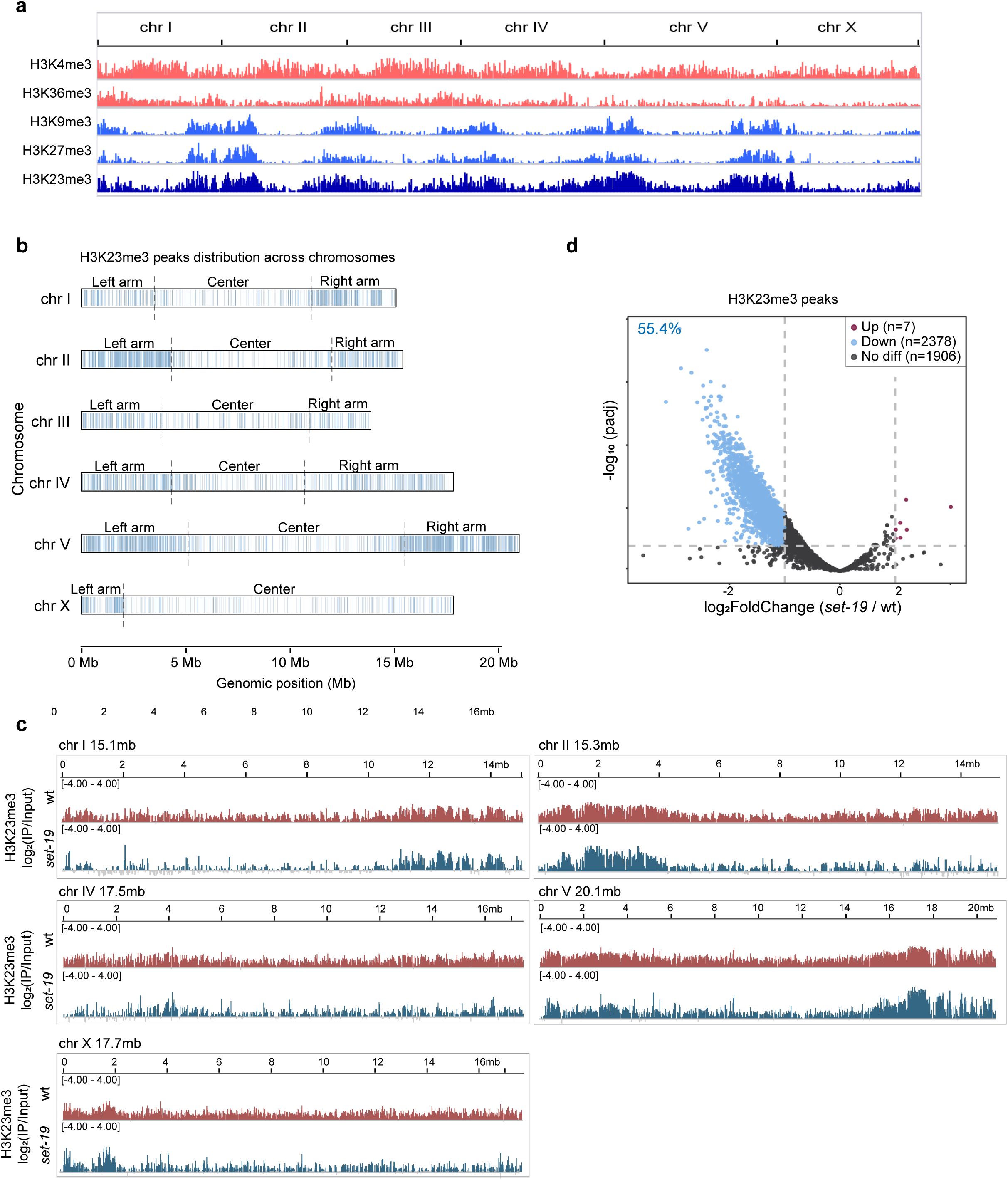
The loss of
set-19
alters genomic H3K23me3 occupancy.
a,
Genome-wide distribution of ChIP-seq peaks for H3K4me3, H3K36me3, H3K9me3, H3K27me3, and H3K23me3, as identified by MACS2.
b,
Distribution of H3K23me3 ChIP-seq peaks across chromosomes in wild-type worms. Peak positions identified by MACS2 from one representative biological replicate (replicate 1) are shown along each chromosome. Chromosome arms and centers are indicated.
c,
Comparison of H3K23me3 ChIP-seq signal distributions across chromosomes in wild-type and
set-19(ok1813)
worms. Mean log₂ enrichment over input is shown (
N
= 2 biological replicates).
d,
Differential H3K23me3 peak analysis between
set-19(ok1813)
and wild-type worms. Volcano plot showing log
2
FoldChange (
set-19
/wt) versus -log
10
(adjusted p value [padj]) from DiffBind
54
analysis. Differentially enriched peaks are highlighted (|log
2
FoldChange|>1 and padj < 0.01).
N
= 2 biological replicates.
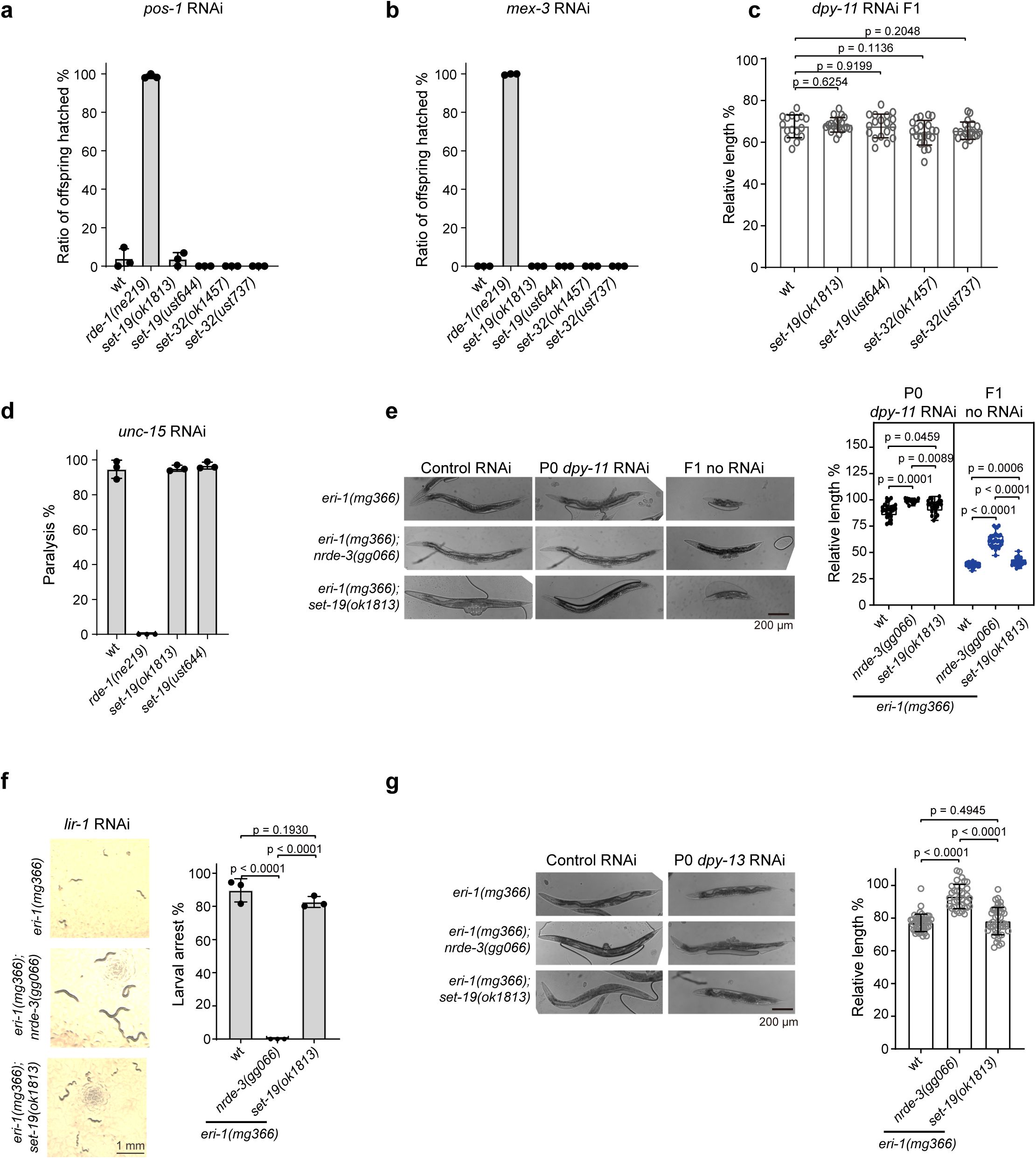
SET-19 is dispensable for feeding RNAi responses.
a, b,
Quantification of embryonic lethality following feeding RNAi targeting
pos-1
(
a
) or
mex-3
(
b
). Synchronized adult hermaphrodites of the indicated genotypes were exposed to dsRNA-expressing bacteria, and the ratio of hatched offspring was scored.
c,
Analysis of RNAi efficiency using
dpy-11
feeding RNAi. The indicated animals were exposed to
dpy-11
dsRNA until adulthood, and the relative body length of F1 progeny was measured. Individual data points represent single animals.
d,
Quantification of paralysis phenotypes following
unc-15
RNAi in the indicated genotypes. The percentage of paralyzed animals was scored.
e,
Animals of the indicated genotypes were exposed to
dpy-11
RNAi in the P0 generation and transferred to control bacteria. Representative images of P0 and F1 animals are shown (left). The relative body length of P0 and F1 animals was quantified (right).
f,
Larval arrest analysis following
lir-1
RNAi in the
eri-1(mg366)
background. Representative images of larvae are shown (left), and the percentage of animals exhibiting larval arrest was quantified (right).
g,
Analysis of the RNAi response to
dpy-13
feeding RNAi in the
eri-1(mg366)
background. The indicated animals were exposed to dsRNA until adulthood, and the relative body length was measured.
Expression pattern of SET-32.
a,
Fluorescence images of L4-stage animals expressing GFP::SET-32.
b,
Subcellular localization of GFP::SET-32 in the germline at different developmental stages.
c,
Subcellular localization of GFP::SET-32 in embryos.
Data availability
ChIP-seq and RNA-seq data have been deposited in the Genome Sequence Archive (GSA) at the National Genomics Data Center (NGDC) under accession number CRA037051. The mass spectrometry proteomics data have been deposited in the iProX repository with the accession codes IPX0015174000 and PXD073043. All data generated or analyzed during this study are included in the manuscript and supporting files. Source data files have been provided for all figures.
Acknowledgements
We are grateful to the members of the Guang laboratory for their comments and suggestions. We are grateful to the International
C. elegans
Gene Knockout Consortium and the National Bioresource Project for providing the strains. Some strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440).
Additional information
Funding
This work was supported by grants from the National Key R&D Program of China (2022YFA1302700), the National Natural Science Foundation of China (32230016, 32270583, 32300438, 32400435, and 32470633), the Research Funds of Center for Advanced Interdisciplinary Science and Biomedicine of IHM (QYPY20230021), and the Fundamental Research Funds for the Central Universities (WK9100250107).
Author contributions
Y.S., M. Huang, X.F. and S.G. conceptualized the research; M.X., X.F. and S.G. designed the research; M.X., Z.F., M. Huang, and C.Y. performed the research; X.C., X. Huang, C.Z., M. Hong and X. Hou contributed new reagents; J.C., X. Hou, S.L. and M.L. provided technical guidance on bioinformatics analysis and protein purification; M.X., X.F. and S.G. wrote the paper.
Funding
MOST | National Key Research and Development Program of China (NKPs) (2022YFA1302700)
Shouhong Guang
MOST | National Natural Science Foundation of China (NSFC) (32230016)
Shouhong Guang
MOST | National Natural Science Foundation of China (NSFC) (32270583)
Chengming Zhu
MOST | National Natural Science Foundation of China (NSFC) (32300438)
Xinya Huang
MOST | National Natural Science Foundation of China (NSFC) (32400435)
Xiangyang Chen
MOST | National Natural Science Foundation of China (NSFC) (32470633)
Xuezhu Feng
MOE | Fundamental Research Funds for the Central Universities (Fundamental Research Fund for the Central Universities) (WK9100250107)
Xiangyang Chen
Additional files
References
1.
Histone post-translational modifications - cause and consequence of genome function
Nat. Rev. Genet
23
:563–580
PubMed
Google Scholar
2.
H3K9me3-heterochromatin loss at protein-coding genes enables developmental lineage specification
Science
363
:294–297
PubMed
Google Scholar
3.
H3K9me selectively blocks transcription factor activity and ensures differentiated tissue integrity
Nat. Cell Biol
23
:1163–1175
PubMed
Google Scholar
4.
Germ line–inherited H3K27me3 restricts enhancer function during maternal-to-zygotic transition
Science
357
:212–216
PubMed
Google Scholar
5.
Histone modifications in stem cell development and their clinical implications
Stem Cell Rep
15
:1196–1205
PubMed
Google Scholar
6.
Epigenetic inheritance: histone bookmarks across generations
Trends Cell Biol
24
:664–674
PubMed
Google Scholar
7.
The chemical biology of reversible lysine post-translational modifications
Cell Chem Biol
27
:953–969
PubMed
Google Scholar
8.
The role of histone modifications: from neurodevelopment to neurodiseases
Signal Transduct Target Ther
7
:217
PubMed
Google Scholar
9.
Dot1p modulates silencing in yeast by methylation of the nucleosome core
Cell
109
:745–756
https://doi.org/10.1016/s0092-8674(02)00759-6
PubMed
Google Scholar
10.
Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase
Cell
112
:711–723
https://doi.org/10.1016/s0092-8674(03)00114-4
PubMed
Google Scholar
11.
SET for life: biochemical activities and biological functions of SET domain-containing proteins
Trends Biochem. Sci
38
:621–639
PubMed
Google Scholar
12.
Sequence analysis of acetylation and methylation in two histone H3 variants of alfalfa
J. Biol. Chem
265
:17157–17161
https://doi.org/10.1016/s0021-9258(17)44882-4
PubMed
Google Scholar
13.
Organismal differences in post-translational modifications in Histones H3 and H4
J. Biol. Chem
282
:7641–7655
PubMed
Google Scholar
14.
Systematic identification of methyllysine-driven interactions for histone and nonhistone targets
J. Proteome Res
9
:5827–5836
PubMed
Google Scholar
15.
A method for systematic mapping of protein lysine methylation identifies functions for HP1β in DNA damage response
Mol. Cell
50
:723–735
PubMed
Google Scholar
16.
Quantitative proteomics reveals that the specific methyltransferases Txr1p and Ezl2p differentially affect the mono-, di- and trimethylation states of Histone H3 lysine 27 (H3K27)
Mol Cell Proteomics
12
:1678–1688
PubMed
Google Scholar
17.
Methylation of histone H3K23 blocks DNA damage in pericentric heterochromatin during meiosis
eLife
3
:e02996
PubMed
Google Scholar
18.
H3K23me2 is a new heterochromatic mark in Caenorhabditis elegans
Nucleic Acids Res
43
:9694–9710
PubMed
Google Scholar
19.
Dynamic changes of histone H3 marks during Caenorhabditis elegans lifecycle revealed by middle-down proteomics
Proteomics
16
:459–464
PubMed
Google Scholar
20.
Reader domain specificity and lysine demethylase-4 family function
Nat. Commun
7
:13387
PubMed
Google Scholar
21.
Chromatin organization at the nuclear periphery as revealed by image analysis of structured illumination microscopy data
J. Cell Sci
130
:2066–2077
PubMed
Google Scholar
22.
JMJD-1.2 controls multiple histone post-translational modifications in germ cells and protects the genome from replication stress
Sci. Rep
8
:3765
PubMed
Google Scholar
23.
Changes of histone H3 lysine 23 acetylation and methylation in porcine somatic cells, oocytes and preimplantation embryos
Theriogenology
148
:162–173
PubMed
Google Scholar
24.
H3K23me1 is an evolutionarily conserved histone modification associated with CG DNA methylation in Arabidopsis
Plant J
90
:293–303
PubMed
Google Scholar
25.
Caenorhabditis elegans nuclear RNAi factor SET-32 deposits the transgenerational histone modification, H3K23me3
eLife
9
:e54309
PubMed
Google Scholar
26.
Two H3K23 histone methyltransferases, SET-32 and SET-21, function synergistically to promote nuclear RNAi-mediated transgenerational epigenetic inheritance in Caenorhabditis elegans
Genetics
229
:iyae206
PubMed
Google Scholar
27.
Amplification of siRNA in Caenorhabditis elegans generates a transgenerational sequence-targeted histone H3 lysine 9 methylation footprint
Nat. Genet
44
:157–164
PubMed
Google Scholar
28.
A nuclear Argonaute promotes multigenerational epigenetic inheritance and germline immortality
Nature
489
:447–451
PubMed
Google Scholar
29.
Decoupling the downstream effects of germline nuclear RNAi reveals that H3K9me3 is dispensable for heritable RNAi and the maintenance of endogenous siRNA-mediated transcriptional silencing in Caenorhabditis elegans
Epigenetics Chromatin
10
:6
PubMed
Google Scholar
30.
The Nrde pathway mediates small-RNA-directed histone H3 lysine 27 trimethylation in Caenorhabditis elegans
Curr. Biol
25
:2398–2403
PubMed
Google Scholar
31.
Chromatin modifiers SET-25 and SET-32 are required for establishment but not long-term maintenance of transgenerational epigenetic inheritance
Cell Rep
25
:2259–2272
PubMed
Google Scholar
32.
C. elegans heterochromatin factor SET-32 plays an essential role in transgenerational establishment of nuclear RNAi-mediated epigenetic silencing
Cell Rep
25
:2273–2284
PubMed
Google Scholar
33.
The RNAi Inheritance Machinery of Caenorhabditis elegans
Genetics
206
:1403–1416
PubMed
Google Scholar
34.
Repressive chromatin in Caenorhabditis elegans: establishment, composition, and function
Genetics
208
:491–511
PubMed
Google Scholar
35.
A role for Set1/MLL-related components in epigenetic regulation of the Caenorhabditis elegans germ line
PLoS Genet
7
:e1001349
PubMed
Google Scholar
36.
The SET-2/SET1 histone H3K4 methyltransferase maintains pluripotency in the Caenorhabditis elegans germline
Cell Rep
9
:443–450
PubMed
Google Scholar
37.
Caenorhabditis elegans chromatin-associated proteins SET-2 and ASH-2 are differentially required for histone H3 Lys 4 methylation in embryos and adult germ cells
Proc. Natl. Acad. Sci. U.S.A
108
:8305–8310
PubMed
Google Scholar
38.
Two SET domain containing genes link epigenetic changes and aging in Caenorhabditis elegans
Aging Cell
11
:315–325
PubMed
Google Scholar
39.
Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery
Cell
150
:934–947
PubMed
Google Scholar
40.
Argonaute NRDE-3 and MBT domain protein LIN-61 redundantly recruit an H3K9me3 HMT to prevent embryonic lethality and transposon expression
Genes Dev
35
:82–101
PubMed
Google Scholar
41.
Differential localization and independent acquisition of the H3K9me2 and H3K9me3 chromatin modifications in the Caenorhabditis elegans adult germ line
PLoS Genet
6
:e1000830
PubMed
Google Scholar
42.
The MES-2/MES-3/MES-6 complex and regulation of histone H3 methylation in C. elegans
Curr. Biol
14
:1639–1643
PubMed
Google Scholar
43.
Trans-generational epigenetic regulation of C. elegans primordial germ cells
Epigenetics Chromatin
3
:15
PubMed
Google Scholar
44.
A histone methylation network regulates transgenerational epigenetic memory in C. elegans
Cell Rep
7
:113–126
PubMed
Google Scholar
45.
A SET domain-containing protein and HCF-1 maintain transgenerational epigenetic memory
Nat. Commun
17
:1462
PubMed
Google Scholar
46.
H3K9me1/2 methylation limits the lifespan of daf-2 mutants in C. elegans
eLife
11
:e74812
PubMed
Google Scholar
47.
The evolving metabolic landscape of chromatin biology and epigenetics
Nat. Rev. Genet
21
:737–753
PubMed
Google Scholar
48.
The impact of one carbon metabolism on histone methylation
Front. Genet
10
:764
PubMed
Google Scholar
49.
Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism
Cell Metab
22
:861–873
PubMed
Google Scholar
50.
Intrinsic catalytic properties of histone H3 lysine-9 methyltransferases preserve monomethylation levels under low S-adenosylmethionine
J. Biol. Chem
299
:104938
PubMed
Google Scholar
51.
Broad chromosomal domains of histone modification patterns in C. elegans
Genome Res
21
:227–236
PubMed
Google Scholar
52.
Recombinational landscape and population genomics of Caenorhabditis elegans
PLoS Genet
5
:e1000419
PubMed
Google Scholar
53.
Meiotic recombination, noncoding DNA and genomic organization in Caenorhabditis elegans
Genetics
141
:159–179
PubMed
Google Scholar
54.
Differential oestrogen receptor binding is associated with clinical outcome in breast cancer
Nature
481
:389–393
PubMed
Google Scholar
55.
H3K9me3 is required for inheritance of small RNAs that target a unique subset of newly evolved genes
eLife
8
:e40448
PubMed
Google Scholar
56.
pos-1 encodes a cytoplasmic zinc-finger protein essential for germline specification in C. elegans
Development
126
:1–11
PubMed
Google Scholar
57.
MEX-3 is a KH domain protein that regulates blastomere identity in early C. elegans embryos
Cell
87
:205–216
https://doi.org/10.1016/s0092-8674(00)81339-2
PubMed
Google Scholar
58.
The rde-1 gene, RNA interference, and transposon silencing in C. elegans
Cell
99
:123–132
https://doi.org/10.1016/s0092-8674(00)81644-x
PubMed
Google Scholar
59.
A novel thioredoxin-like protein encoded by the C. elegans dpy-11 gene is required for body and sensory organ morphogenesis
Development
129
:1185–1194
PubMed
Google Scholar
60.
An Argonaute transports siRNAs from the cytoplasm to the nucleus
Science
321
:537–541
PubMed
Google Scholar
61.
Genetic organization of the region around UNC-15 (I), a gene affecting paramyosin in Caenorhabditis elegans
Genetics
96
:639–648
PubMed
Google Scholar
62.
RDE-1 slicer activity is required only for passenger-strand cleavage during RNAi in Caenorhabditis elegans
Nat. Struct. Mol. Biol
16
:207–211
PubMed
Google Scholar
63.
dpy-13: a nematode collagen gene that affects body shape
Cell
55
:567–576
https://doi.org/10.1016/0092-8674(88)90215-2
PubMed
Google Scholar
64.
RNA interference can target pre-mRNA: consequences for gene expression in a Caenorhabditis elegans operon
Genetics
153
:1245–1256
PubMed
Google Scholar
65.
Comprehensive single-cell transcriptional profiling of a multicellular organism
Science
357
:661–667
PubMed
Google Scholar
66.
Methylation and demethylation activities of a C. elegans MLL-like complex attenuate RAS signalling
Developmental Biology
341
:142–153
PubMed
Google Scholar
67.
Two conserved epigenetic regulators prevent healthy ageing
Nature
579
:118–122
PubMed
Google Scholar
68.
Muscle-specific histone H3K36 dimethyltransferase SET-18 shortens lifespan of Caenorhabditis elegans by repressing daf-16a expression
Cell Rep
22
:2716–2729
PubMed
Google Scholar
69.
The histone H3K36 methyltransferase MES-4 acts epigenetically to transmit the memory of germline gene expression to progeny
PLoS Genetics
6
:e1001091
PubMed
Google Scholar
70.
Argonaute slicing is Required for heterochromatic silencing and spreading
Science
313
:1134–1137
PubMed
Google Scholar
71.
Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi
Science
297
:1833–1837
PubMed
Google Scholar
72.
Nuclear RNAi maintains heritable gene silencing in Caenorhabditis elegans
Proc. Natl. Acad. Sci. U.S.A
108
:19683–19688
PubMed
Google Scholar
73.
piRNAs can trigger a multigenerational epigenetic memory in the germline of C. elegans
Cell
150
:88–99
PubMed
Google Scholar
74.
The spatial dynamics of tissue-specific promoters during C. elegans development
Genes Dev
24
:766–782
PubMed
Google Scholar
75.
Histone H3K9 methylation is dispensable for Caenorhabditis elegans development but suppresses RNA:DNA hybrid-associated repeat instability
Nat. Genet
48
:1385–1395
PubMed
Google Scholar
76.
Dual sgRNA-directed gene knockout using CRISPR/Cas9 technology in Caenorhabditis elegans
Sci. Rep
4
:7581
PubMed
Google Scholar
77.
Extraction, purification and analysis of histones
Nat. Protoc
2
:1445–1457
PubMed
Google Scholar
78.
Chemical derivatization of histones for facilitated analysis by mass spectrometry
Nat. Protoc
2
:933–938
PubMed
Google Scholar
79.
EpiProfile 2.0: A computational platform for processing epi-proteomics mass spectrometry data
J. Proteome Res
17
:2533–2541
PubMed
Google Scholar
80.
Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans
Gene
263
:103–112
https://doi.org/10.1016/s0378-1119(00)00579-5
PubMed
Google Scholar
81.
Systematic functional analysis of the Caenorhabditis elegans genome using RNAi
Nature
421
:231–237
PubMed
Google Scholar
82.
Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype
Nat. Biotechnol
37
:907–915
PubMed
Google Scholar
83.
featureCounts: an efficient general purpose program for assigning sequence reads to genomic features
Bioinformatics
30
:923–930
PubMed
Google Scholar
84.
Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2
Genome Biol
15
:550
PubMed
Google Scholar
85.
Differential chromatin marking of introns and expressed exons by H3K36me3
Nat. Genet
41
:376–381
PubMed
Google Scholar
86.
Systematic characterization of chromodomain proteins reveals an H3K9me1/2 reader regulating aging in C. elegans
Nat. Commun
14
:1254
PubMed
Google Scholar
87.
Fast gapped-read alignment with Bowtie 2
Nat. Methods
9
:357–359
PubMed
Google Scholar
88.
The Sequence Alignment/Map format and SAMtools
Bioinformatics
25
:2078–2079
PubMed
Google Scholar
89.
Model-based analysis of ChIP-Seq (MACS)
Genome Biology
9
:R137
PubMed
Google Scholar
90.
Integrative Genomics Viewer
Nat. Biotechnol.
29
:24–26
PubMed
Google Scholar
91.
ChIPseqSpikeInFree: a ChIP-seq normalization approach to reveal global changes in histone modifications without spike-in
Bioinformatics
36
:1270–1272
PubMed
Google Scholar
Identification of a somatic H3K23me3 methyltransferase SET-19 in C. elegans
Genome Sequence Archive
ID CRA037051
Identification of a somatic H3K23me3 methyltransferase SET-19 in C. elegans
iProX
ID IPX0015174000
Identification of a somatic H3K23me3 methyltransferase SET-19 in C. elegans
iProX
ID IPX0015174000
Chromatin accessibility dynamics across C. elegans development and ageing
NCBI Sequence Read Archive
ID SRR7164162
A team of heterochromatin factors collaborates with small RNA pathways to combat repetitive elements and germline stress
NCBI Sequence Read Archive
ID SRR5297105
Chromatin accessibility dynamics across C. elegans development and ageing
NCBI Sequence Read Archive
ID SRR7164198
Chromatin accessibility dynamics across C. elegans development and ageing
NCBI Sequence Read Archive
ID SRR7164186
Article and author information
Author information
Cite all versions
You can cite all versions using the DOI
10.7554/eLife.111326
. This DOI represents all versions, and will always resolve to the latest one.
Copyright
© 2026,
Xu et al.
This article is distributed under the terms of the
Creative Commons Attribution License
, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
views
0
downloads
0
citations
0
Views, downloads and citations are aggregated across all versions of this paper published by eLife.





