 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库Environmental temperature is a strong driver of subspecies competition in themicrobiome
Abstract
Most microbiome research focusses on the taxonomic composition at the species level to understand the impact of environmental factors, but intraspecific diversity has largely been ignored. To address this significant knowledge gap, we took advantage of the simple, culturable microbiome of
Drosophila
. First, we documented that natural populations of
D. simulans
harbor three diverged clades of
Lactiplantibacillus plantarum
, a key nutritional symbiont. We studied the distinct ecological roles of these three clades by exposing flies with their native microbiome to two temperature regimes in the laboratory. Tracking the three clades within the complete
Drosophila
microbiome over a period of more than 10 years at two temperatures, we identified strikingly distinct dynamics in response to the selection regime. We confirmed the functional differentiation of the three clades using
in vitro
growth dynamics and
in vivo
mono-association assays. Our results highlight that environmental selection operates at the subspecies level. Therefore, we conclude that the functional diversification of the microbiome can only be understood when intra– and interspecific diversity is considered.
Introduction
Intraspecific diversity has largely been overlooked in microbial ecology surveys. This can be in part attributed to the success of 16S rRNA profiling, which allows the characterization of species diversity with a very high efficiency (
1
,
2
), but cannot identify intraspecific diversity (
3
). Although environmental variation has been identified as one of the major factors driving species composition, its role for intraspecific adaptation has been largely overlooked (
3
). This blind spot of microbiome research is particularly surprising as it is known that conspecific strains, i.e. strains from the same species, can display large phenotypic variation (
4
). Clinically relevant species like
Escherichia coli
, in which different subspecies can be either pathogenic or commensal are a particularly illustrative example (
5
).
Although a few studies in environmental microbial communities have provided evidence for the functional importance of intraspecific variation (
6
,
7
), such analyses are considerably more difficult. Therefore, descriptive correlative studies are more common than functional assays (
8
). In the gut-dwelling bacterium
B. fragilis
conspecific strains compete for the same niche leading to the mutual exclusion of toxicogenic and non-toxicogenic strains in the gut environment (
9
). Not only in the human gut, but also in lakes, soil, and the sea conspecific strains frequently co-occur in sympatry (
10
–
12
). It has even been even reported that strain richness, i.e. sympatric strain diversity within the same species, stabilizes microbial communities in fluctuating environments (
13
,
14
).
In this work, we further explored the functional implications of intraspecific diversity within the same microbial population. We focused on the species
Lactiplantibacillus plantarum
, an extremely versatile lactic acid bacterium that has been associated with a variety of different habitats, including plants, the gastro-intestinal tracts of mammals and insects, as well as food such as meat, dairy, and pickled products (
15
).
L. plantarum
is a facultative symbiont of
Drosophila
(
16
–
19
).
Drosophila
larvae can enhance the growth of
L. plantarum,
which in response releases essential nutrients for the larvae under poor diet conditions (
18
). As a result of this nutritional symbiosis,
L. plantarum
is a prevalent taxon in the
Drosophila
microbiome (
17
).
To study the implications of intraspecific diversity of
L. plantarum
in fruit flies, we took advantage of an experimentally evolved
Drosophila simulans
population that has been adapting to two novel temperature regimes, hot and cold, for more than ten years. We detected three clades of
L. plantarum
co-occurring within the native microbiome of
D. simulans
population before the experimental temperature was modified. However, the exposure of the fly population to the novel temperature regimes altered the intraspecific composition of
L. plantarum,
revealing functional differences between the co-occurring clades. These functional differences were experimentally validated
in vitro
and
in vivo
. Our results provide a clear example for functional diversification of intraspecific diversity and demonstrate how experimental evolution can uncover this otherwise hidden functional diversity.
Results
Genome sequencing reveals the presence of three
L. plantarum
clades in the experimentally evolving populations
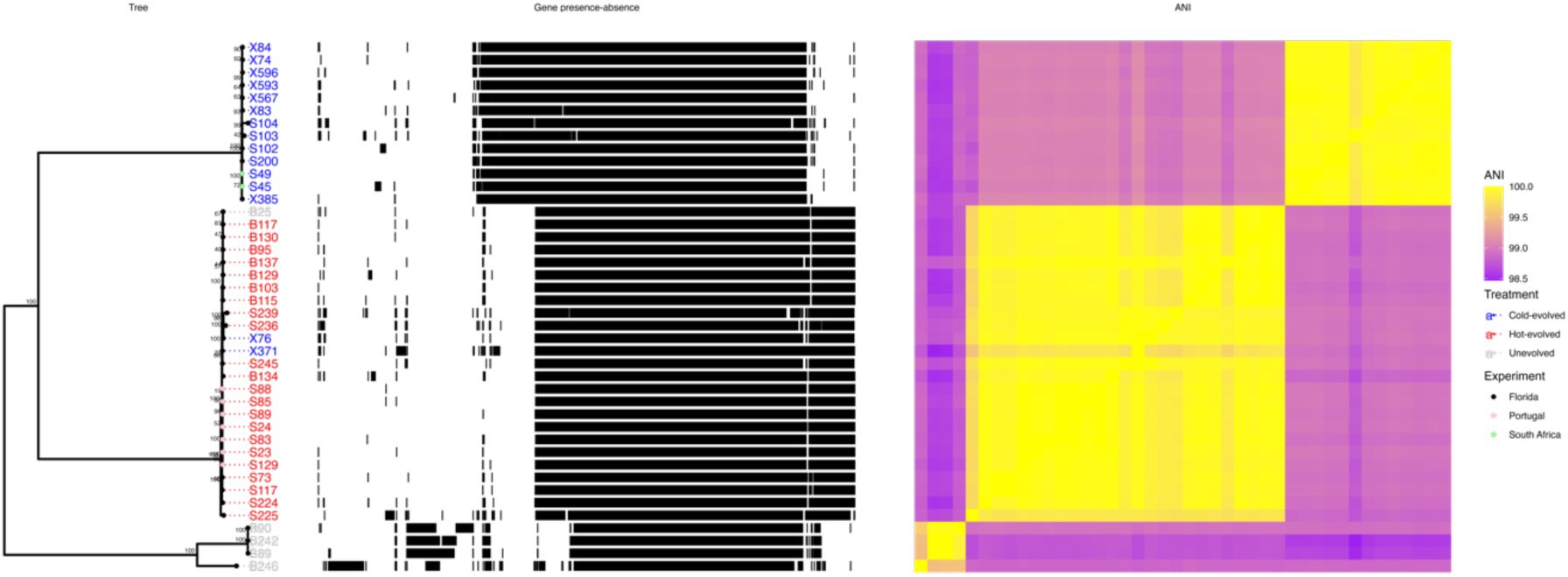
We sampled 33 isolates from the Florida experiment (See Materials and Methods for details) to characterize the diversity of
L. plantarum
(
Figure 1
,
Table S1
). The isolates formed three clades based on the pairwise average nucleotide identity (ANI). Between-group ANI values were 98.4-99.17%, below the ∼99.5% threshold for conspecific strain identity (
20
). On the other hand, the identity within groups was 99.48-99.99% (
Figure 2
). The clustering was consistent irrespective of whether we used SNPs in the core genome or presence/absence patterns of accessory genes. This indicates that the three clades are old with very limited genetic exchange.
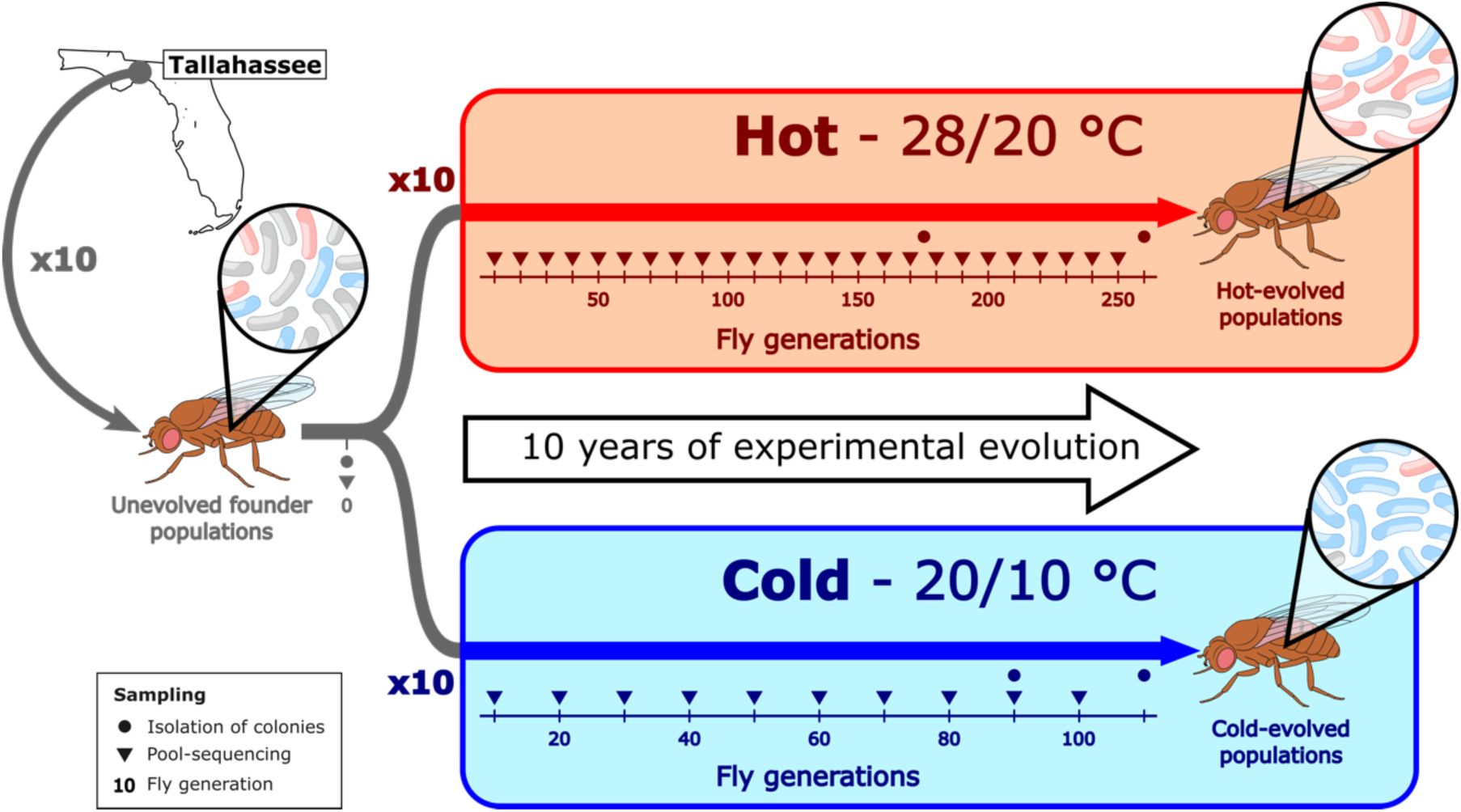
Experimental setup to study the long-term evolution of
L. plantarum
in
Drosophila simulans
populations.
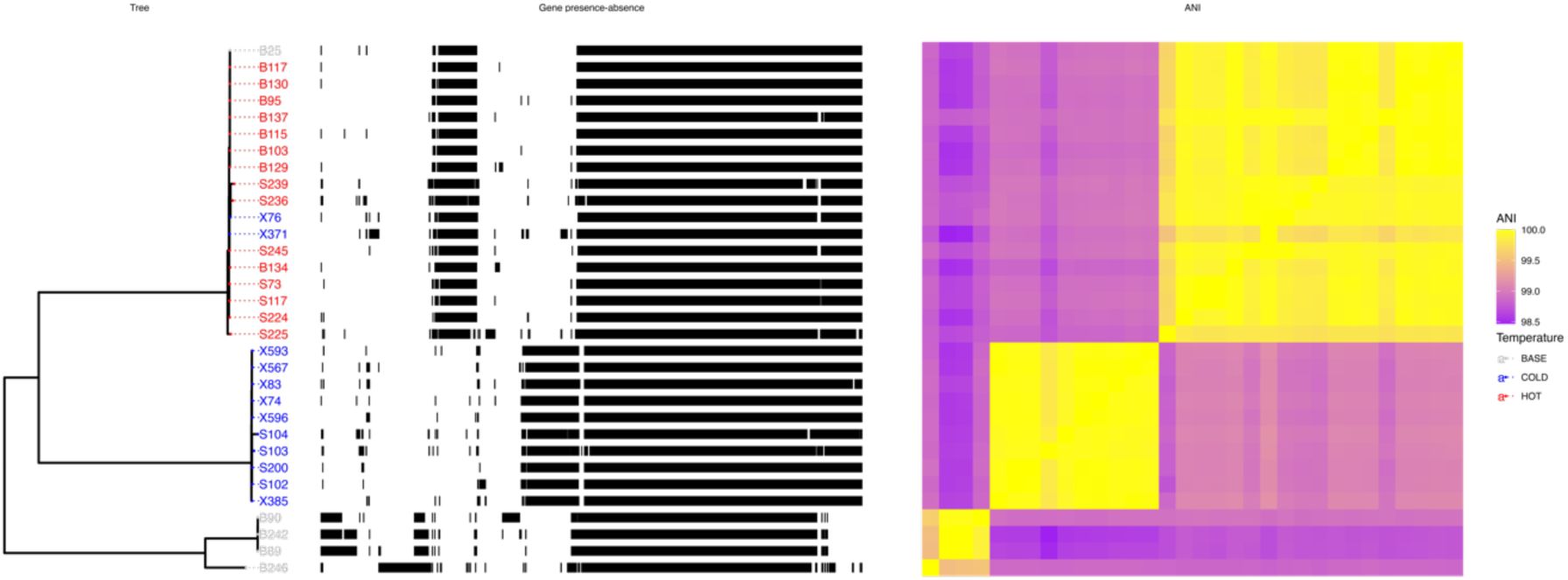
Pangenome of
L. plantarum
genomes isolated from the Florida experiment (
Table S1
), sorted by phylogeny.
Left panel: Maximum likelihood tree built based on the core genome alignment. Tree leaves correspond to the name of the isolate and are coloured based on isolation origin: ancestral population (grey), hot-evolved population (red) or cold-evolved population (blue). Numbers at nodes represent bootstrap support values based on 1000 replicates. Middle panel: Pattern of genes presence (black)/absence (white) in each genome. Right panel: pairwise average nucleotide identity between the isolates. The colour ranges between 98.5% (purple) and 100% (yellow).
Since the isolates were sampled either from the unevolved, hot-evolved or cold-evolved populations (
Figure 1
), we tested whether the clades were randomly distributed across the experimental treatments and observed a non-random distribution (Fisher’s exact test, 3×3 contingency table, p = 5.5e-10). In combination with the clustering of clades (
Figure 2
), we conclude that each of the three clades is more prevalent either in the novel experimental populations or in the unevolved founder population, at the beginning of the experiment. We therefore coined the terms H (hot-evolved), C (cold-evolved), and U (unevolved) clades, that will be used throughout the manuscript.
In order to further investigate the association between
L. plantarum
genotype and temperature, we sampled from two additional experiments, obtaining seven isolates from hot-evolved
Drosophila simulans
Portugal and two isolates from constant-cold-evolved
Drosophila simulans
South Africa (
Table S1
). In a phylogenetic tree, the hot-evolved isolates and the constant-cold-evolved isolates grouped into clades H and C, respectively (
Figure S1
). These results do not only support the association between
L. plantarum
clade composition and environmental temperature (Fisher’s exact test, 3×3 contingency table, p = 2.6e-12), but also suggest that clade C is favored irrespective of whether the temperature is fluctuating (20 °C during the day, 10 °C at night) or constant (15 °C). Furthermore, the grouping of isolates originating from different experiments to the same clades indicates that these are not restricted to a single natural
D. simulans
population, but represent global diversity.
The three sympatric clades independently adapted to their host
Our analyses suggest that all three clades are shared among natural
D. simulans
populations, but it is not clear whether they diverged in
D. simulans
or independently colonized their
Drosophila
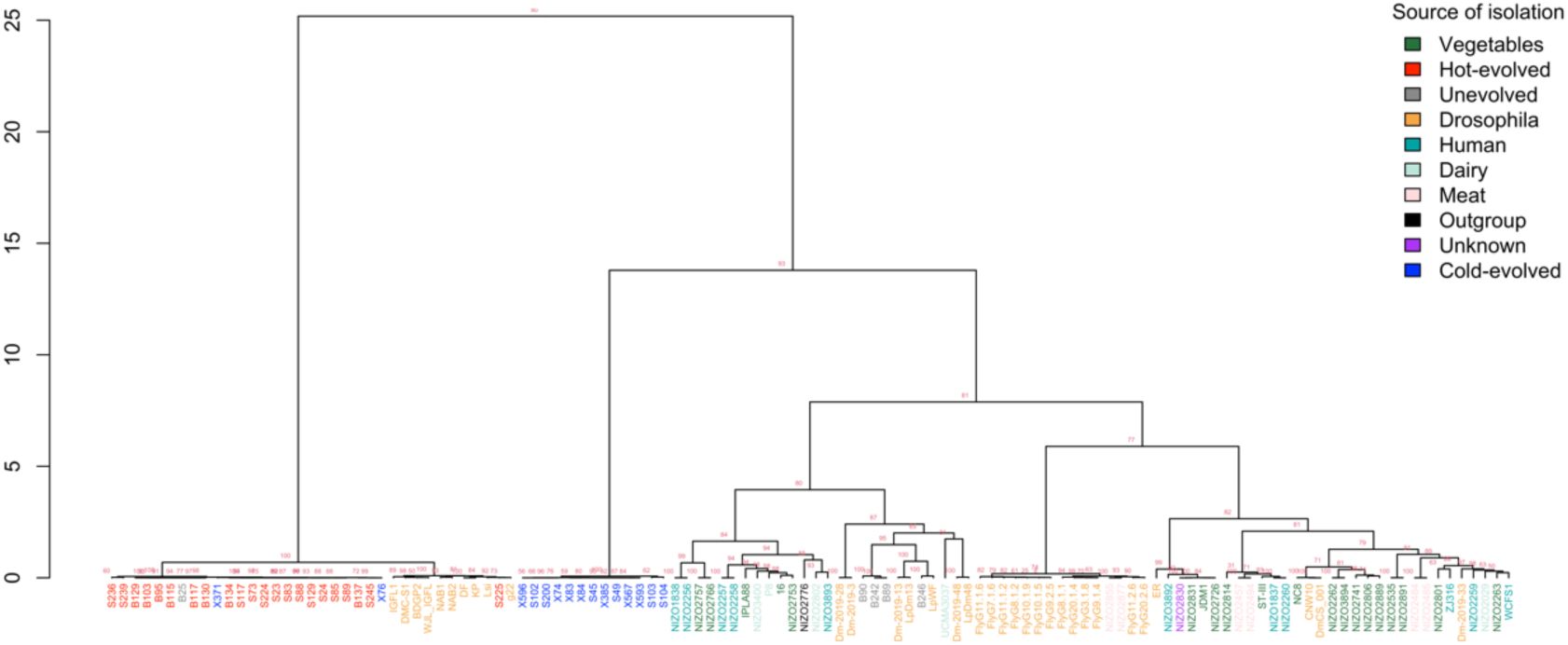
host. We used 79 publicly available
L. plantarum
genomes from several environments and geographic locations (
Table S2
) to reconstruct the phylogeny based on the core genome.
All three
L. plantarum
clades are more closely-related to genomes from other sources than to each other, which implies that they diverged before colonizing
D. simulans
(
Figure 3
). Moreover, the genomes from the clade H are almost identical to public sequences isolated from global collections of
Drosophila melanogaster
(
Figure 3
,
Table S2
). The clade U isolate B246 is closely related to the
Drosophila
-associated isolate LpWF originating from a wild
D. melanogaster
individual (
21
). Clade C did not have a close relative in the collection. The high similarity of the
L. plantarum
sequences isolated from
D. melanogaster
and
D. simulans
highlights that both
Drosophila
species share this component of the microbiome. Most likely a deeper sampling of the
D. melanogaster
microbiome will also detect clade C.
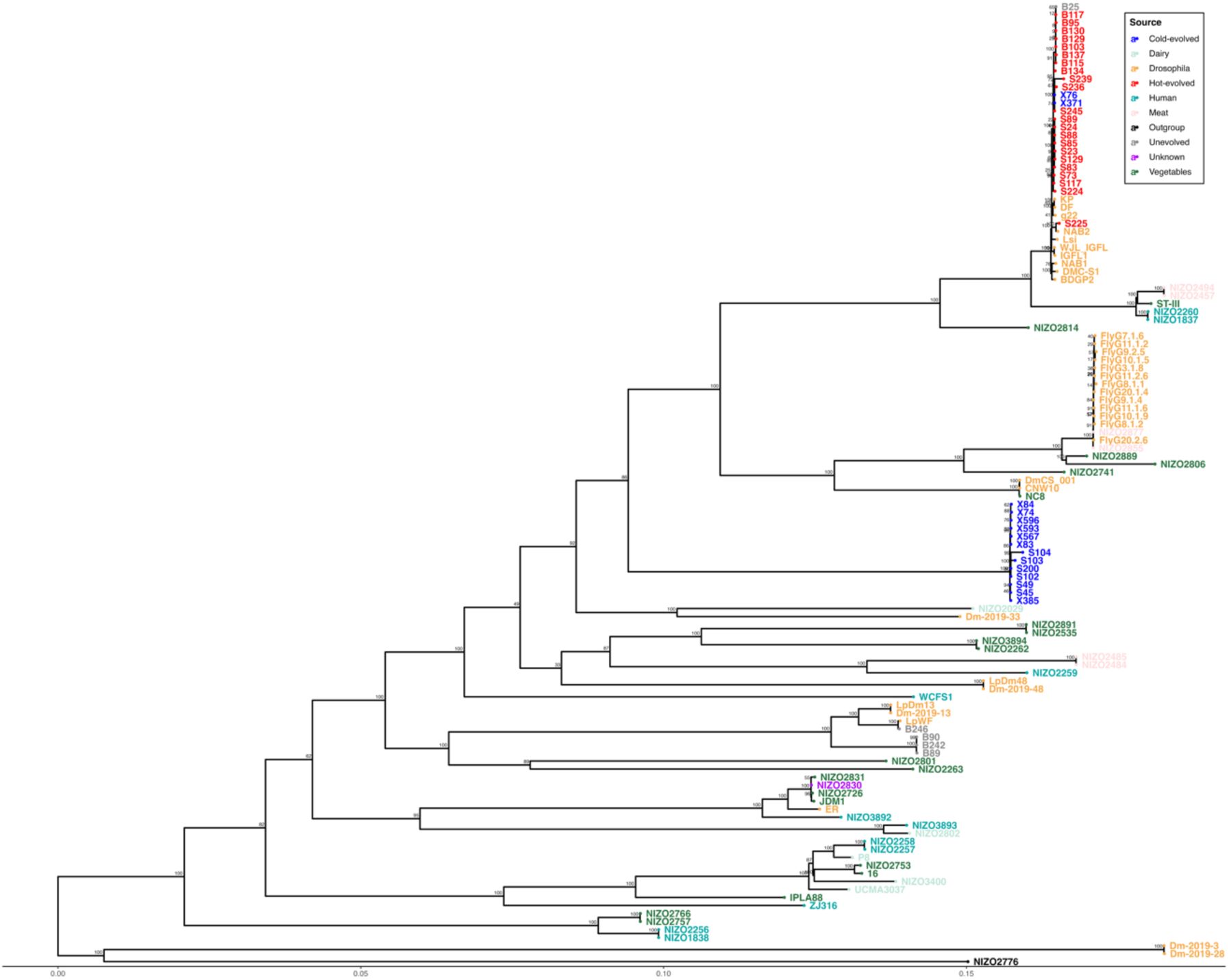
Maximum likelihood tree of 92
L. plantarum
genomes.
Tree leaves are coloured by source of isolation. The genomes from our lab are coloured in according to experimental treatment: unevolved in grey, hot-evolved in red, and cold-evolved in blue. Numbers at nodes represent bootstrap support values based on 1000 replicates.
Despite being phylogenetically divergent, the three sympatric clades could have converged functionally in the process of adaptation to a common environment. In order to test this, we built the
L. plantarum
pangenome including all the available isolates and performed hierarchical clustering based on the presence/absence of accessory genes. We found no grouping of the three sympatric clades (
Figure S2
), which suggests that they are not only phylogenetically diverged, but have the potential to be functionally different based on their pool of accessory genes.
Clade dynamics are temperature-specific and consistent between population replicates
Up to now, our analyses were restricted to isolates sampled at specific time points of the experiment, but since genomic DNA from flies with their microbiome was sequenced throughout the entire experiment (
Figure 1
), a metagenomic analysis could track the frequency trajectories of the three clades to shed more light on the adaptation process. We calculated the proportion of reads assigned to each clade in the reads from the population at a given time point. At the beginning of the experiment, the populations were dominated by clade U (54.5% relative abundance on average), followed by clade C (31.9%), and then clade H (13.6%) (
Figure 4
). However, when the fly populations were subjected to the experimental treatments, clade H became dominant under the hot regime and clade C became dominant under the cold regime. Clade U greatly decreased in relative abundance in both regimes. This suggests that changes in environmental temperature alter the fitness landscape of the population and lead to a new adaptive state, in which the relative fitness of the clades change.
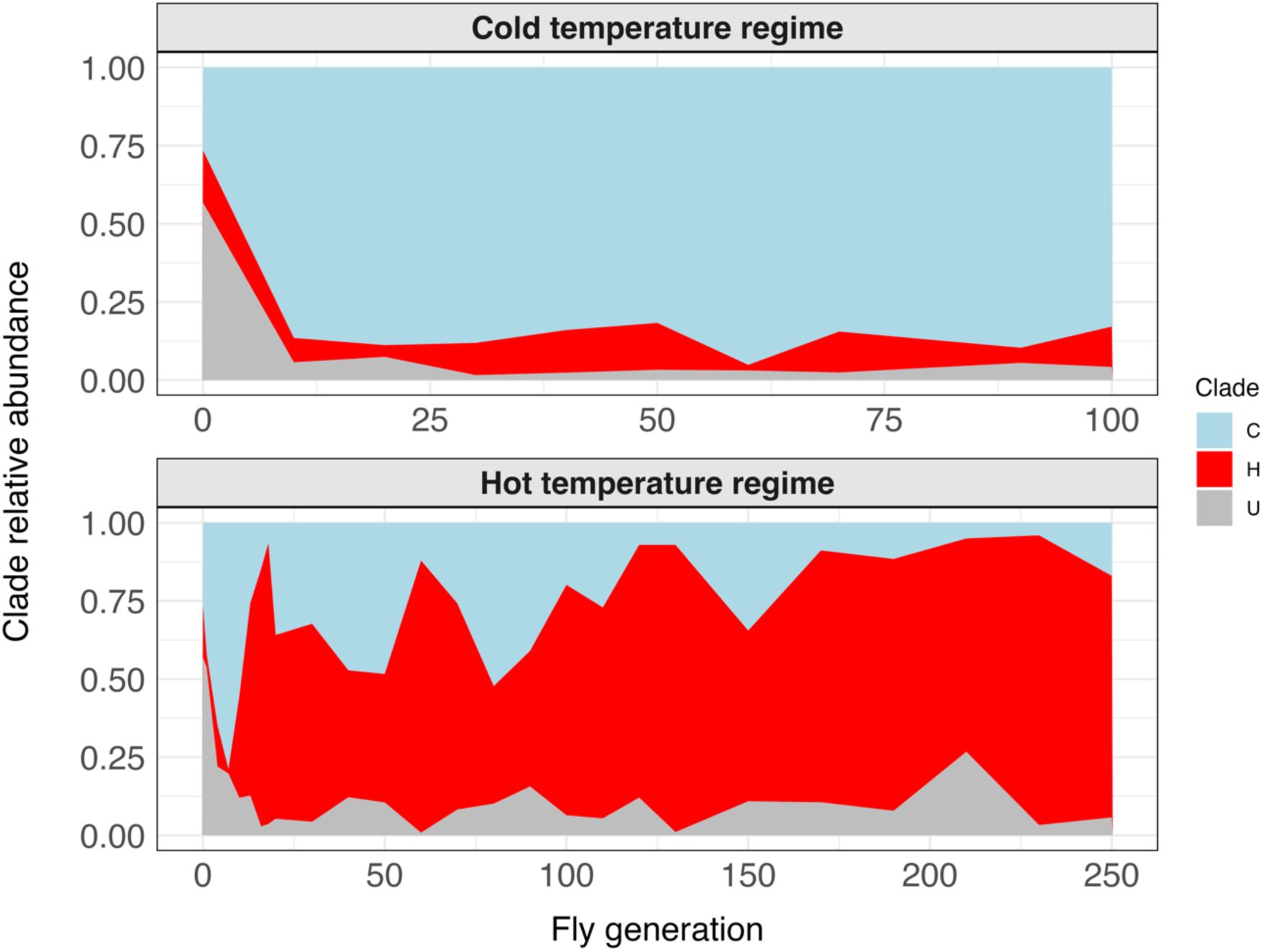
Clade composition over time in each temperature regime depicted as the relative abundance of each
L. plantarum
clade (top: cold, bottom: hot).
Clade C in light blue, clade H in red and clade U in grey. For each temperature regime we summed the reads from the ten replicate populations.
A strong asset of the experimental evolution set up is the availability of 10 replicate populations that were generated from the same founder population and independently maintained throughout the entire experiment under the same conditions. Hence, the comparison of replicates provides an estimate for the strength of deterministic and stochastic forces during the experiment. In the cold regime, all ten replicates exhibited a rapid takeover of clade C (
Figure S3
). In contrast, the hot-evolved replicates were more variable in the rate at which clade H increased in abundance. The most extreme cases were replicates 8 and 10, in which clade C dominated the population for 90 generations before being replaced by clade H (
Figure S3
). Thus, the populations subjected to the hot regime took longer to reach a new equilibrium, which is also less reproducible than that of the cold regime. Overall, the temperature-specific dynamic is consistent across replicates in both regimes, confirming the association between environmental temperature and clade frequency that was observed during the isolation of the clones (
Figures 1
,
2
and
S1
).
Phenotypic differences in liquid culture
The consistent temperature-specific dynamics of
L. plantarum
in the replicate populations resulted in a different clade dominating each novel temperature regime. The fitness differences may arise from an increased growth rate at a given temperature. Alternatively, traits such as toxin-antitoxin systems or host interaction may cause temperature-specific fitness (
22
). To determine if the observed clade dynamics were driven by differences in temperature-dependent growth rates, we measured the growth rate of 29 isolates from the three clades at hot temperature conditions. These isolates represented all sampling events across the two temperature regimes (hot and cold) as well as the unevolved population (
Table S1
). The three groups of isolates differed significantly in growth rate, carrying capacity, and inflection time, i.e. the time it takes an isolate to reach the log phase (
Figure 5
; Kruskal-Wallis test, p < 0.001 for all parameters). Isolates belonging to clade C took on average more than 12 h longer to reach the log growth phase. Clade H had the highest growth rate, followed by U, and C. Clade U reached the highest density, followed by H, and C.
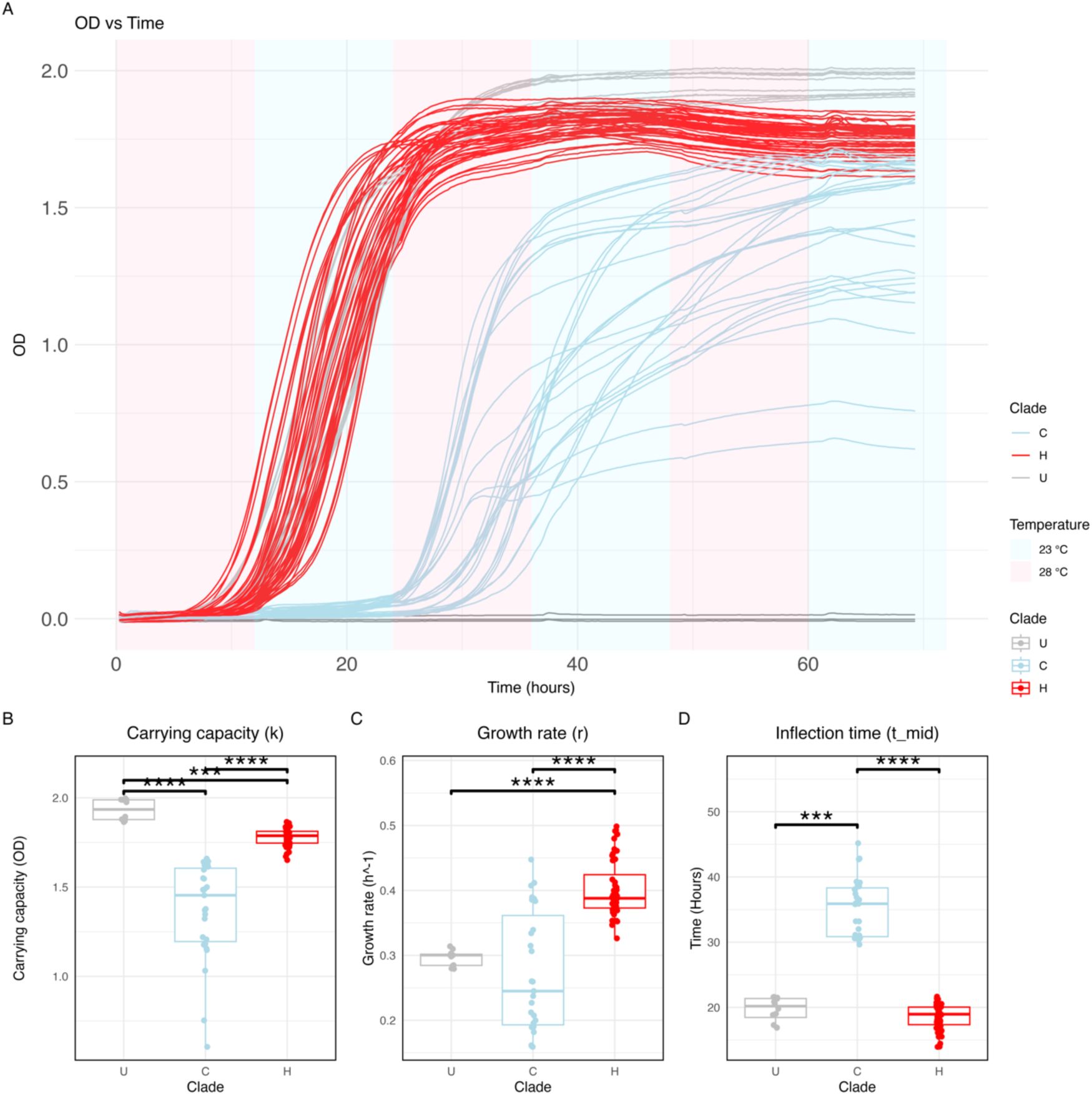
Clade-specific growth dynamics.
(A)
L. plantarum
growth curves overlapped. Lines are coloured by isolate’s clade. Background colour represents the growth temperature in the fluctuating environment. (B-D) Boxplots depicting the carrying capacity, growth rate, and inflection time of each clade. Each dot corresponds to a technical replicate. Data is grouped and coloured by clade. Statistical significance was determined using Dunn’s test with Holm-adjusted p-values. Only significant comparisons are indicated. ****
p
< 0.0001; ***
p
< 0.001; **
p
< 0.01; *
p
< 0.05.
These results reveal the functional diversification of the three clades, but do not support the hypothesis that temperature-specific growth rates caused the shifts in composition. In liquid culture, clade U outperformed clade C in growth rate, carrying capacity, and inflection time despite the latter clade prevailed in the
Drosophila
populations for much longer (
Figure 4
,
Figure S3
). Nevertheless, we caution that the growth conditions in the
Drosophila
environment are radically different to the growth measured in liquid MRS (
23
). Furthermore, inside the fly gut
L. plantarum
lives in a complex community in which the different clades interact with each other, with the other components of the microbiome, and finally also with the host. Bacteria can persist in the community by thriving in the food or by stably colonizing the fly gut. The former strategy might be controlled by growth rate and carrying capacity, but the latter is affected by additional factors such as death rate in the gut and defecation rate, that cannot be assessed in liquid medium (
21
). Moreover, the resource availability is different in the solid fly food and in liquid MRS medium. Thus, observed differences in growth could indicate different strategies to persist in the community.
Fitness effects of
L. plantarum
strains on their host
Although growth dynamics differ significantly among the three clades in liquid culture, they do not explain the observed changes in abundance during experimental evolution. Therefore, we turned to the
Drosophila
host to explore the fitness consequences of colonization by each clade.
L. plantarum
has been shown to increase larval fitness of
Drosophila melanogaster
relative to germ-free flies (
18
,
24
,
25
). We hypothesized that the fitness advantage conferred by
L. plantarum
to its host could depend on the genotype of the symbiont and the environmental temperature, favoring a different clade in each regime. To test this, we designed a set of inoculation experiments in both focal temperatures using two host species,
D. melanogaster
and
D. simulans
, and the three
L. plantarum
clades. While we could produce axenic
D. melanogaster
, attempts to produce axenic
D. simulans
were unsuccessful. For this reason, inoculation experiments involving
D. simulans
were conducted using conventionally reared flies.
In axenic
D. melanogaster
none of the
L. plantarum
clades provided a fitness advantage to the host relative to germ-free controls, contrary to the effects reported in the literature. In the case of clades U and H, the number of offspring and developmental time did not differ significantly from the germ-free control (
Figure 6
and
S6
; Dunn’s test, p > 1 for all pairs). This discrepancy could be explained by the supply of essential amino acids during larval growth by
L. plantarum
, (
18
,
24
–
26
). The high content of dried yeast 24.3 g/l in the fly food used in our experiment likely provided already sufficient amounts of essential amino acids, which negated the growth-promoting effects of
L. plantarum
. Even more surprising was the significantly reduced fitness in the presence of clade C in both temperature regimes (
Figure 6
; Dunn’s test, p < 0.05 for all significant comparisons). A mixture of all three clades resulted in a similar loss in fitness. The fitness reduction was stronger in the first transfer of flies, likely due to a higher bacterial load. This finding suggests an inverse relationship between bacterial abundance and host fitness.
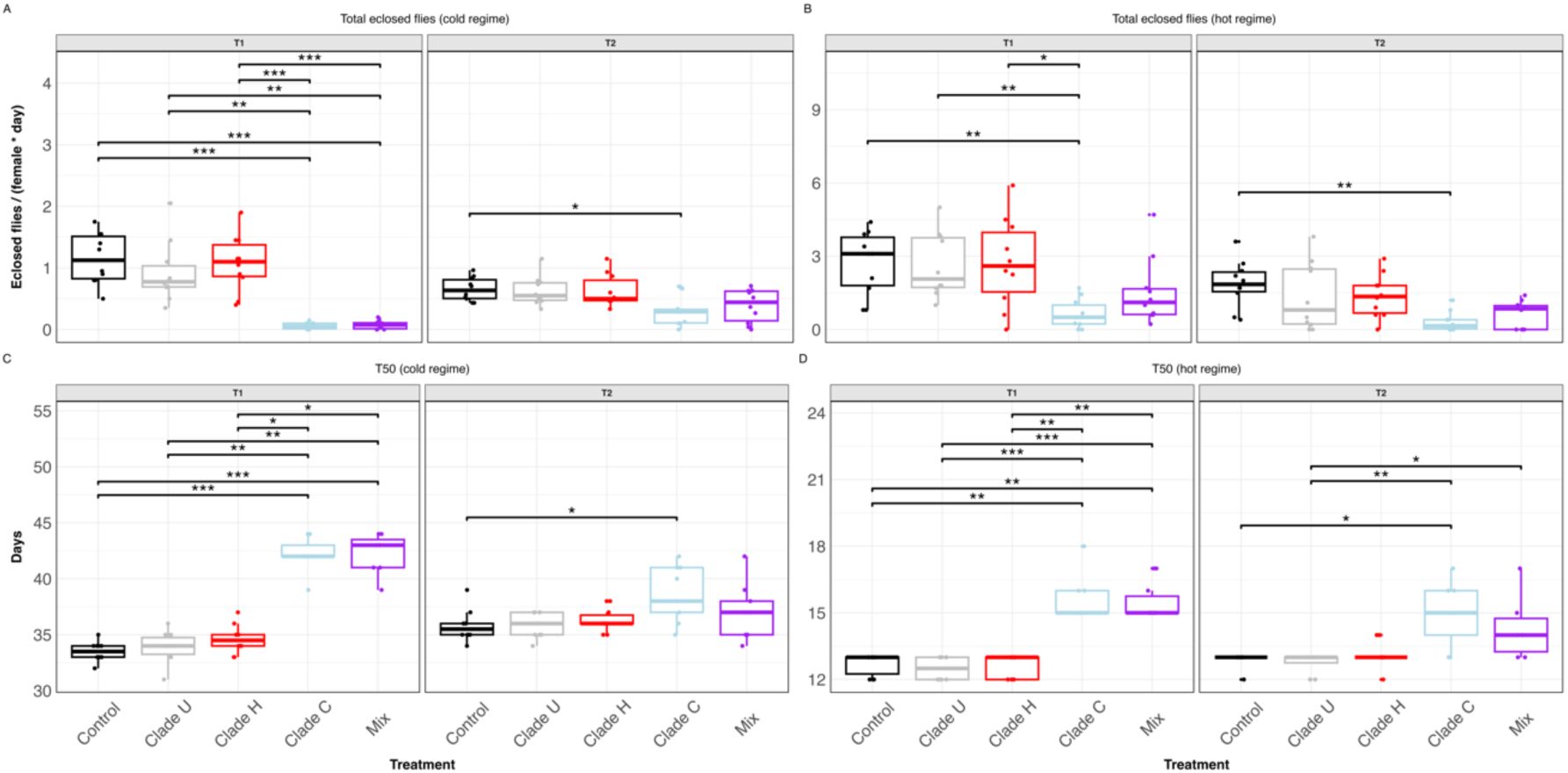
Fitness effect of
L. plantarum
inoculation in axenic
D. melanogaster
.
(A, B) Total number of F1 flies eclosed normalised by day and female under the cold (A) and hot (B) regime. (C, D) Developmental time, estimated as the number of days it takes 50% of the offspring to eclose. Measurements were grouped by inoculation treatment and transfer. Each dot corresponds to a biological replicate (n = 10) Statistical significance was determined using Dunn’s test with Holm-adjusted p-values. Only significant comparisons are indicated. ****
p
< 0.0001; ***
p
< 0.001; **
p
< 0.01; *
p
< 0.05.
In conventionally reared
D. simulans
we did not detect differences in the number of eclosed flies (
Supplementary figure S6
; ANOVA). However, the developmental time was significantly extended after inoculation with clade C at cold temperature (Dunn’s test, p < 0.05 05 for all significant comparisons). We attribute the weaker effects in the non-axenic flies to a reduced
L.
plantarum load due to the presence of other taxa (
21
), or to higher-order interactions with other members of the microbiome (
27
–
29
).
In summary, we observed that clade C, which is dominant in the cold-evolved populations, decreases host fitness when axenic flies are inoculated. The magnitude of this effect varies depending on the environmental temperature, the bacterial load, and the presence of other microbial taxa. Clade-specific effects of
L. plantarum
on the
D. melanogaster
host have been reported (
19
). In a low-protein diet, strains that were not isolated from
Drosophila
, enhanced larval growth relative to germ-free individuals, whereas another
Drosophila-
associated strain did not have any effect. Even a reduction of
Drosophila
lifespan has been reported in mono-association with
L. plantarum
(
30
), but the reduction of adult viability and developmental rate relative to the germ-free control is a novel finding in our study. We can conclude that the outcome of the
L. plantarum
-
Drosophila
symbiosis is clade-specific and can be detrimental to the host.
Functional divergence on the genomic level
Our analyses clearly indicate functional divergence between the three clades at genetic and phenotypic levels (see
Figures S2
and
4
-
6
). Therefore, we were interested in identifying the genes that contribute to this functional differentiation. A comparative KEGG pathway analysis shows that clade C contains the most KEGG Orthologs (KOs), 1163 KOs, followed by clade H (1157 KOs), and clade U (1131 KOs). A subset of 111 orthologs is unique to one or two clades. Of these, 41 were shared by clades H and C (
Figure S7
). Among these differential orthologs fell into 53 KEGG metabolic pathways.
Interestingly, we found a larger repertoire of sugar-related genes in the two lab-selected clades, C and H, than in clade U (
Table S3
,
Figure S7
). One of these genes encodes the enzyme hexosaminidase (EC: ) and confers clades C and H the ability to use chitobiose as carbon source. This oligomer is the main product of chitin degradation, that makes up the flies’ peritrophic matrix and exoskeleton (
31
). Therefore, we speculate that the ability to exploit this ubiquitous source of carbon and nitrogen in the lab-maintained fruit flies, could be a strong target of selection in the lab environment. This could explain why clade U, which displayed a high growth rate and carrying capacity in liquid culture (
Figure 5
), is rapidly outcompeted in the fly populations (
Figures 4
and
S3
).
Furthermore, clade H and C (
Table S3
) harbor different sugar-related functions, indicating that, despite their overlapping functions, these strains possess distinct sugar metabolic capacities. We also identified functional differences in cofactor biosynthesis and respiratory metabolism between clades. These differences do not explain the clade-specific selection, but reflect the different evolutionary histories of the clades (
Table S3
,
figures S8
-
S11
). A detailed description of the functional diversification can be found in the supplement.
Discussion
Intraspecific diversity within the same environment has been reported to be ecologically important to maintain the stability of the entire microbial community (
11
,
13
,
14
). This parameter has been studied in lakes, soils, seas, and the human gut (
10
–
12
). However, little attention has been given to intraspecific variation in
Drosophila
, and insect microbiomes in general (
8
). Based on our results in experimentally evolved fruit flies, we propose that within-species competition, thus far largely overlooked, could contribute to ecological adaptation and evolution of the host, as it has been observed at the species level in
Drosophila
(
32
) and other animals (
33
–
35
).
Our finding of three co-occurring subspecies of
L. plantarum
in our populations is particularly surprising, given the low microbiome richness of
Drosophila
at species level, which is estimated to be orders of magnitude lower than in humans (
36
,
37
). However, the intraspecific richness of
L. plantarum
in our flies was three times higher than that estimated in human gut microbiomes (
11
). This discrepancy between inter– and intra-species diversity suggests that the
Drosophila
microbiome is more complex than previously thought (
16
,
27
,
38
). However, this diversity exists below the species level and has therefore remained cryptic due to the limitations of amplicon sequencing (
3
).
By altering the experimental temperature at which the populations were maintained, we discovered consistent shifts in the composition of
L. plantarum
subspecies, that inevitably led to the dominance of a single clade. Further inoculation experiments revealed that clade composition significantly impacts the fitness of the host.
L. plantarum
genotypes differ in the fitness advantage that they provide to the host (
19
). However, to our knowledge, no
L. plantarum
genotype has yet been reported to negatively affect
Drosophila
development. Our finding shows that the well-characterized nutritional symbiosis between
Drosophila
and
L. plantarum
depends on the bacterial genotype and cannot be generalized to the entire species. Furthermore, clade C outcompeted the other two clades in the cold populations despite negatively affecting the host. This demonstrates that the benefits of parasitism or mutualism in the
Drosophila
microbiome are context-dependent, as already observed in humans (
39
).
Finally, we also gained insight into how the microbiome of the fly could have adapted to laboratory conditions. Unlike the non-selected clade U, the two lab-selected clades, C and H, had the unique genetic potential to exploit the byproducts of chitin degradation. Our results align with previous research on the “domestication” of the
Drosophila
microbiome. Comparative genomics in
Acetobacter
have revealed that bacteria associated with lab-reared
Drosophila
were selected for the presence of uricase genes and the loss of flagella, relative to their wild-isolated counterparts (
40
). In
Acetobacter
, the ability to degrade uric acid excreted by the host provides a significant competitive advantage (
40
). We hypothesize that the same may apply to the ability to metabolize host-derived chitin. We conclude that our findings are consistent with laboratory adaptation, and extends previous observations from
Acetobacter
to
Lactiplantibacillus
.
Overall, our work emphasizes the importance of intraspecific diversity in maintaining the stability of microbial communities and the host’s ability to adapt to environmental changes. The current climate crisis has sparked a significant interest in how environmental temperature affects microbial communities at the species level (
41
–
43
). Our study highlights that even subspecies diversity plays a key role in adaptation to environmental temperature, and probably other abiotic factors. Therefore, we propose that a comprehensive understanding of adaptive processes and ecological dynamics in the case of host-microbiome interactions depends critically on including diversity at the subspecies level.
Materials and Methods
Experimental evolution
Twenty replicate populations were set up using 202 isofemale lines from a natural
D. simulans
population collected in Florida (
44
). The populations were maintained in a 12 h photoperiod and at two different novel temperature regimes: ten replicates were kept in a fluctuating hot environment (28 °C during the day and 18 °C at night), whereas the other ten were kept in a fluctuating cold environment (20 °C during the day and 10 °C at night). The census population size of the replicates was 1000-1250 with a 50:50 sex ratio. The flies in each replicate were equally distributed across five 300 ml bottles containing 60 ml of standard
Drosophila
medium (300 g Agar + 990 g sugar beet syrup + 1000 g malt syrup + 2,310 g corn flour + 390 g soy flour + 900 g yeast in 37.5 L water) (
45
). The evolved
D. simulans
populations were sequenced in 10 generation intervals using Pool-Seq (
46
,
47
). These sequencing reads have been used to study the evolutionary dynamics of fruit flies (
47
). Similar pool-seq time series data have been previously used to analyze long-term endosymbiont and microbiome dynamics in
D. melanogaster
populations evolving in the same experimental conditions (
48
–
50
).
We also isolated
L. plantarum
from two other experimental evolution experiments, one originated from a South African population reared at constant 15 °C and the other from Portugal, which was reared in the fluctuating hot regime as described earlier (
51
). Both experimental evolution studies followed the same culturing regime that was used for the Florida populations, with temperature being the only experimentally altered variable (
52
).
Bacterial isolation
We primarily obtained data from the experimental evolution studies using the Florida
D. simulans
founder population and collected 33
L. plantarum
genomes from five different time points; two time points from each experimental temperature and one time point corresponding to the ancestral flies (
Table S1
). In addition, we obtained
L. plantarum
genomes from two other experimental evolution studies. Two genomes from the South African experiment and seven genomes from the Portugal experiment.
Around 100 flies from each population were homogenized in sterile PBS using an autoclaved pestle. The homogenate was diluted to avoid physical contact between colonies and streaked on agar plates with three different media: De Man, Rogosa, and Sharpe (MRS), mannitol and tryptic soy. The plates were incubated at 28 °C for 48 h. We identified
L. plantarum
using PCR with custom
Lactiplantibacillus
-specific primers (in 5’-3’ orientation, LacF:
GATGGGCGCTTACCCGATTA
, LacR:
CTGCCCCGCAAATTGTTTCA
). The proportion of
L. plantarum
colonies in the plates, relative to other taxa, varied across growth media and fly populations. Colonies which could successfully be amplified were picked and regrown to stationary phase in the same medium from which they were isolated, but in liquid format. A glycerol stock of each isolate was made by mixing the culture with glycerol (50% v/v) in 1:1 proportion.
DNA extraction and sequencing
Genomic DNA was extracted from all the replicate populations using the high salt method (
53
). The fly populations were sampled in 10 generations intervals starting from generation 0. At sampling the age of the flies varied between four and eight days for the hot environment and between nine and 16 days for the cold environment. Pools of flies were sequenced at various time points, using a range of library kits, insert sizes, and read lengths (
47
).
For sequencing,
the L. plantarum
isolates were grown to stationary phase in MRS medium. gDNA was isolated with the high salt extraction method (
53
). Additionally, a lysozyme pre-treatment was used to degrade Gram-positive cell wall (
54
). Briefly, the pellet was resuspended in 480 µl of EDTA 50 mM. The suspension was treated with 120 µl of lysozyme (20 mg/ml dissolved in NaCl 30 mM – EDTA 2mM) and incubated for two hours at 37 °C. After this, the samples were centrifuged at maximum speed, the supernatant was discarded, and the pellet was further treated following the high salt gDNA extraction. Genomic DNA was quality-controlled on an Agilent Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA) and subsequently used to prepare DNBSEQ short-read libraries, and 2×150bp reads were sequenced on a DNBSEQ-G400 (MGI Tech Co., Ltd., Shenzhen, China).
Genomic assemblies
Sequencing reads were processed using BBDuk (
BBMap
, 2025 v38.90) to remove adapters, ΦX174 sequences and low-quality bases with the following parameters: ktrim=r, k=23, mink=11, hdist=1, tbo, tpe. Reads were assembled using SPAdes v4 (
56
) with kmer sizes 21, 33, 55, 77, 99, and 127. Subsequently, CheckM2 v1.0.1 (
57
) was used to estimate the completeness and contamination of each assembly, and the taxonomy was determined with GTDB-Tk v2.1.1 (
58
). Ten assemblies had more than 5% contamination, probably due to accidentally picking two adjacent colonies. We used Centrifuge v1.0.4 (
59
) to classify and exclude contigs with exogenous origin from these assemblies.
Comparative genomics and phylogeny
We built the pangenome and phylogenetic tree of the collection of genomes isolated from our lab (
Table S1
). First, we used Prokka v1.14.6 (
60
) to predict the genes in all the genomes. Roary v3.13 (
61
) was used to construct the isolates’ pangenome and extract the aligned genes belonging to the core genome, i.e. those that are present in all genomes. SNP-sites v2.5.1 (
62
) was used to extract the variable positions from the concatenated core genes. A maximum likelihood tree of the aforementioned variable positions was built using the model GTR+ASC and 1000 bootstraps with IQ-TREE v3.0.1 (
63
). Additionally, pairwise average nucleotide identity between all the isolates was computed using the program PyANI v (
64
), with ANIb as alignment method using the whole genomes. This workflow was carried out for the subset of 33 genomes isolated from the Florida experiment and for the whole dataset of 42 genomes, including those isolated from South Africa and Portugal experiments. Unless specified otherwise, subsequent analyses were conducted using the whole dataset pangenome.
The same phylogenetic and pangenomic methods were used for the extended
L. plantarum
genomes collection, including 79 publicly available genomes (
Table S2
). In order to root the tree, an additional tree was made including the reference genome from the sister species
Lactiplantibacillus paraplantarum
(GCA_003641145.1) as outgroup. The
L. plantarum
isolate that was most closely related to
L. paraplantarum
, NIZO2776, was used to root the tree.
In order to investigate which metabolic functions were differentially encoded in each clade, we followed the Anvi’o v7.1 pangenomic workflow (
65
,
66
) to identify “gene clusters” that are unique and ones that are shared across the genomes isolated from our lab. These gene clusters were further annotated against the KEGG database using the built-in Anvi’o function ‘anvi-run-kegg-kofams’. The script ‘anvi-compute-functional-enrichment’ (
67
) was then used to screen for KEGG functions that are differentially enriched between the clades. Briefly, this script estimates the fraction of genomes from each clade that encode each KEGG Ortholog (KO) and tests whether they are significantly associated to one or more groups. We used the R package ggKegg v1.4.1 (
68
) to visualize the KEGG pathways in which at least one gene was enriched.
Competitive mapping and clade relative abundance calculation
First, we pre-filtered the
Drosophila
pool-seq reads by removing the reads that have eukaryotic or
Wolbachia
origin. We used Bowtie v2.5.4 (
69
) with stringent settings (-D 500 –R 40 –N 0 –L 20 –I S,1,0.50 –-no-mixed –-no-discordant) to map each pool-seq data set against a collection of reference genomes that included
D. simulans
(GCF_016746395.2),
D. melanogaster
(GCF_000001215.4),
D. mauritiana
(GCF_004382145.1),
H. sapiens
(GRCh38),
M. musculus
(GCF_000001635.27),
A. thaliana
(GCF_000001735.4),
S. cerevisiae
(GCF_000146045.2),
C. lupus
(GCF_011100685.1) and a several insect-infecting
Wolbachia
strains (GCF_000008025.1, GCF_000022285.1, GCF_000376585.1, GCF_000376605.1, GCF_000475015.1). We kept the read pairs in which none of the reads mapped to any of the genomes in the collection, which should predominantly represent prokaryotic sequences.
Then, we used SuperPang v0.9.6 (
70
) to build the reference graph pangenome of each
L. plantarum
clade using all the genomes belonging to the clade as input (
Table S1
,
Figure 3
). In order to calculate the clades relative abundance in each pre-filtered pool-seq, we used BBMap (
BBMap
, 2025 v38.90) with “perfect mode” settings to competitively map each read against the three concatenated clade pangenomes. For each sample, the number of reads that mapped uniquely to each clade was counted and normalized to Reads Per Million (RPM) in order to account for differences in genome size and sample depth.
We benchmarked our approach to confirm the relative abundance estimates from our competitive mapping pipeline. Briefly, we simulated 200k HiSeq reads from each reference graph pangenome using InSilicoSeq v2.0.1 (
71
), and used ‘seqkit sampl’ v2.3.0 (
72
) to mix them in different ratios, maintaining the original size of 200k reads: 1:0:0, 0:1:0, 0:0:1, 1:1:1, 2:1:7, and 8:1:1. Each of the combined read sets was randomly subsampled from 150k to 10 reads (10, 20, 40, 60, 80, 100, 200, 300, 400, 500, 600, 700, 800, 900, 1k, 2k, 5k, 10k, 150k reads) and mapped competitively to the three clades. We confirmed that the relative abundances were correctly estimated with as few as 100-1000 reads uniquely mapping the references (
Figure S4
).
Growth assays
The glycerol stocks from all the available
L. plantarum
isolates were re-grown in agar MRS. A single colony per isolate was grown in liquid MRS and passaged daily for five days in order to remove the influence of freezing. On the fifth day, each suspension was normalized to an optical density (OD
600
) of 0.01, and serially diluted 1:100 in MRS. We selected 31 isolates that represented samples from all the available time points and temperature regimes, and plated them in triplicates in a flat-bottom 96-wells plate. Three wells were filled with sterile MRS medium as blank controls. Later, we detected contamination in two of the isolates, leaving us with 29 isolates. The plate was incubated for 72 h and OD
600
was measured every 15 minutes in a Synergy H1 plate reader. The temperature regime was chosen to be similar to the hot conditions of the fly populations, but due to limitations of the spectrophotometer’s cooling capacity instead of 28 °C during the day and 18 °C at night, we set it to 28 °C during the day and 23 °C at night. Orbital shaking was set to 425 cpm. The R package GrowthCurver v0.3.1 (
73
) was used to infer the growth parameters from the growth curves. All statistical analyses were conducted in R v4.4.2 (
74
) using the package rstatix v0.7.2 (
75
). We used the Kruskal-Wallis test to assess overall differences in growth parameters among the clades. For post-hoc pairwise comparisons, Dunn’s test was applied, with p-value adjustments based on the Holm method. Differences were considered significant at p < 0.05.
Production of axenic flies
We generated germ-free flies, using a modified version of the dechorionation protocol described by Kietz
et al.
(
76
). Specifically, we used 2.8% active chlorine bleach instead of 1% and supplemented bleach, EtOH, and deionized H
2
O with TritonX (1% v/v). The addition of this detergent prevents dechorionated eggs from sticking to the walls of the tubes, thus facilitating their transfer to bottles. The whole process was carried out in sterile conditions using a laminar flow hood. Axenic flies were maintained via periodic transfers to new sterile food inside the laminar flow hood. We first carried out this procedure with several
D. simulans
isofemale lines, but it was not possible to fully eliminate the residual microbiome despite several rounds of treatment. However, we could produce and maintain axenic
D. melanogaster
Oregon-R flies. For this reason, we decided to use
D. melanogaster
as axenic host in the inoculation experiments.
Inoculation experiments
A representative isolate from each clade (
Table S1
, B89 for ancestral, S103 for cold, and S239 for hot) was re-grown from the glycerol stocks in MRS medium. After three transfers, OD
600
was normalized to 0.05. Autoclaved vials with axenic fly food were inoculated with 50 µl of each bacterial suspension (10 vials per treatment). In total five treatments were used: the pure culture from each genotype, a mixture of the three genotypes in equal proportions (“mix”), and control, in which the vials were inoculated with 50 µl of fresh MRS medium. After overnight absorption of the liquid, twenty axenic flies (ten female and ten male flies) were added to each vial and allowed to lay eggs for either 24 h (hot conditions) or 48 h (cold conditions). After this step, the flies were transferred to a new set of vials with axenic, uninoculated food, and allowed to lay eggs for another 24 h (hot conditions) or 72 h (cold conditions). The adults were discarded and the vials were incubated at hot (28 °C during the day and 18 °C at night) or cold conditions (20 °C during the day and 10 °C at night) until the new generation eclosed. The number of eclosed flies was recorded daily. We estimated the total number of flies per vial and the developmental time as proxies of fitness. We calculated developmental time as the day from the start of the experiment until the day at which 50% of the flies in the vial were eclosed. The inoculation experiment was carried out with axenic
D. melanogaster
Oregon-R and non-axenic
D. simulans
from Florida. We used the Kruskal-Wallis test to assess overall differences in the two fitness phenotypes among the treatments. For post-hoc pairwise comparisons, Dunn’s test was applied, with p-values were adjusted using the Holm method. Differences were considered significant at p < 0.05.
Supporting Information
Extended clade-specific differences in KEGG metabolic pathways
Clades C and H encode a shared genetic repertoire related to sugar metabolism that is lacking in clade U. In contrast, the latter does not encode any unique sugar-related function. Clade U is unable to hydrolyze chitobiose into N-acetyl-glucosamine monomers because it lacks the enzyme hexosaminidase (EC: ). It also lacks transaldolase activity (EC: ), that connects the Embden–Meyerhof–Parnass pathway with the non-oxidative pentose phosphate pathway (
77
). Finally, it cannot use sorbitol as a carbon source, since it does not encode the PTS transporter to internalize it (EC: ;
Supplementary figure S8
) nor the enzyme sorbitol dehydrogenase (EC: ), that converts sorbitol into fructose. These metabolic capacities, specially chitobiose degradation, could play a role in the rapid decrease in abundance of clade U observed in both temperature regimes (
Supplementary Table S3
). Chitobiose is the main product of chitin degradation, that makes up the flies’ exoskeleton (
78
). We have detected the presence of microbial chitinases in the metagenomic reads (unpublished observation), which suggests that chitobiose might be available in the community. Thus, the ability to exploit this ubiquitous source of carbon and nitrogen could be very advantageous in the fly microbiome context, but would not affect the fitness in liquid culture (
Figures 4
and
5
). We also found other sugar-related functions are differentially present between clades C and H (
Supplementary Table S3
), suggesting that, despite having overlapping functions, these clades also encode unique sugar-related functions.
We also found differences in cofactor biosynthesis pathways. Although all the analyzed genomes can import riboflavin, the capacity to produce it
de novo
is enriched in clades C and H (
Supplementary Table S3
;
Supplementary figure S9
). This vitamin is essential in many physiological processes (
79
). Therefore, the possibility to synthetize it
de novo
under low extracellular riboflavin conditions is an advantage. In contrast, clade U has the unique capacity to synthesize
de novo
guanylyl molybdenum cofactor (
Supplementary figure S10
), that is essential in molybdenum-dependent enzymes (
80
). One of such enzymes is the nitrate reductase system Nar. Interestingly, clade U also harbors the operons
nreABC
and
narGHIJ
, that encode for genes that sense anoxic conditions and use nitrate as terminal electron donor instead of oxygen, respectively (
81
–
83
). The presence of this molybden-dependent alternative respiratory system exclusively in clade U suggests a different evolutionary history, in which this clade was exposed to anaerobic conditions before adapting to
Drosophila
(
Supplementary figure S11
). Similarly, the enzyme sulfur oxidoreductase (EC: ) is highly enriched in clade C, which suggests that it can potentially use sulfur as terminal electron acceptor in absence of oxygen (
Supplementary figure S11
).
Among the genes that were uniquely present in clade C, we found
sbnA
and
sbnB
, that mediate the synthesis of L-2,3-diaminopropionic acid (
84
). This unusual amino acid serves as precursor of antibiotics and siderophores (
84
), but the rest of the biosynthesis pathways are absent in the genomes. However, by itself is also a potent enzymatic inhibitor (
85
,
86
). Although it goes beyond the scope of this work, we hypothesize that the synthesis of this compound by clade C could be responsible for the observed dose-dependent toxicity upon inoculation in the axenic host (
Figure 6
). As a sanity check, we blasted the two protein sequences against the clustered non-redundant NCBI database. The best matches had identities of maximum 60%, and belonged to
Streptococcus
,
Bacillus
and
Chitinophaga
. No publicly available
L. plantarum
encodes these genes, which further supports the exceptionality of clade C observed in the phylogeny (
Figure 3
).
Supplementary Figures
Pangenome of all L. plantarum genomes (
Table S1
), sorted by phylogeny.
Left panel: Maximum likelihood tree built based on the core genome alignment. Tree leaves correspond to the name of the isolate and are coloured based on isolation origin: unevolved populations (grey), hot-evolved populations (red) or cold-evolved populations (blue). Tree tips are coloured based on the experiment from which the isolate was obtained: Florida (black), Portugal (pink) or South Africa (green). Numbers at nodes represent bootstrap support values based on 1000 replicates. Middle panel: Pattern of genes presence (black)/absence (white) in each genome. Right panel: heat map depicting the pairwise average nucleotide identity between the isolates. The colour ranges between 98.5% (purple) and 100% (yellow).
Hierarchical clustering of 92
L. plantarum
genomes based on the presence-absence of accessory genes.
Tree leaves are coloured by source of isolation. The genomes from our lab are coloured in according to experimental treatment: unevolved in grey, hot-evolved in red, and cold-evolved in blue. Red values in each branch indicate the p-value for each cluster’s robustness based on 1000 bootstrap iterations.
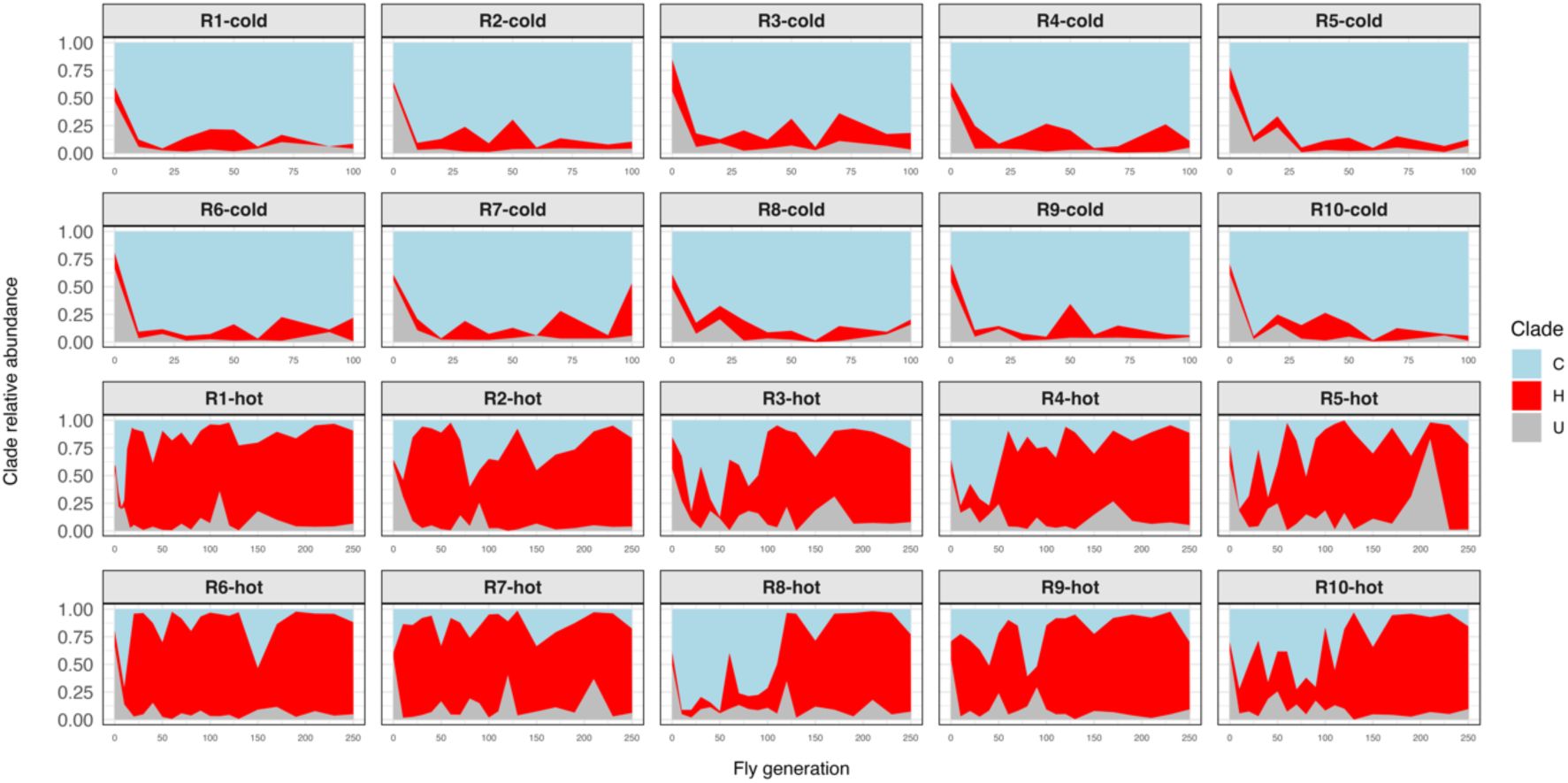
Clade composition over time per replicate population and temperature regime depicted as the relative abundance of each L. plantarum clade.
Clade C in light blue, clade H in red and clade U in grey.
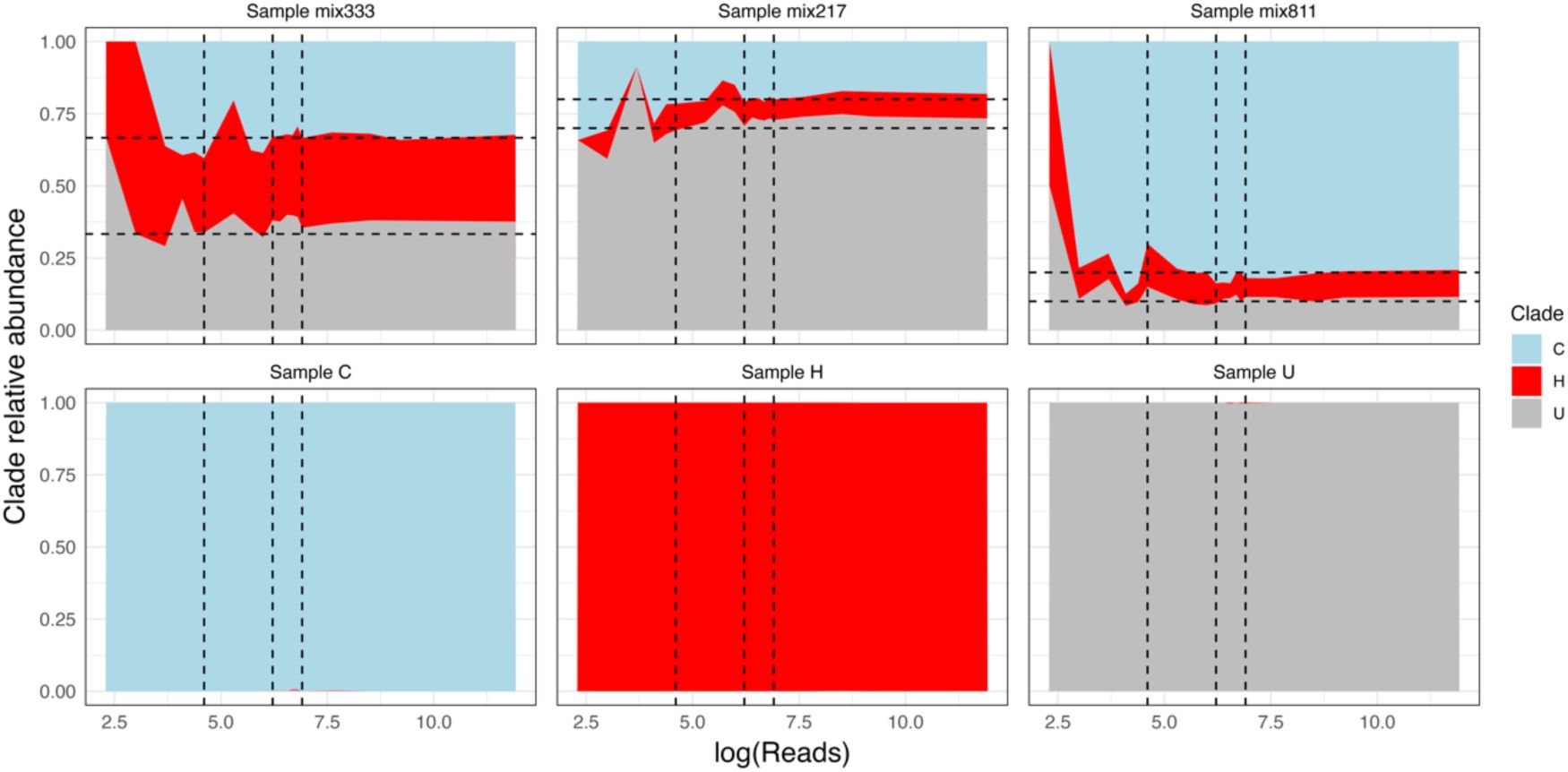
Relative abundance of each L. plantarum clade across different reads depths in simulated reads sets with known clade composition.
Dashed vertical lines show the values of log(100), log(500), and log(1000) reads. Dashed horizontal lines show the true simulated relative abundances of each clade in the reads set. The top row titles reference to the ratio of reads from each clade following descending order: C:H:U. The bottom plots depict scenarios in which all the reads belong to a single taxon.
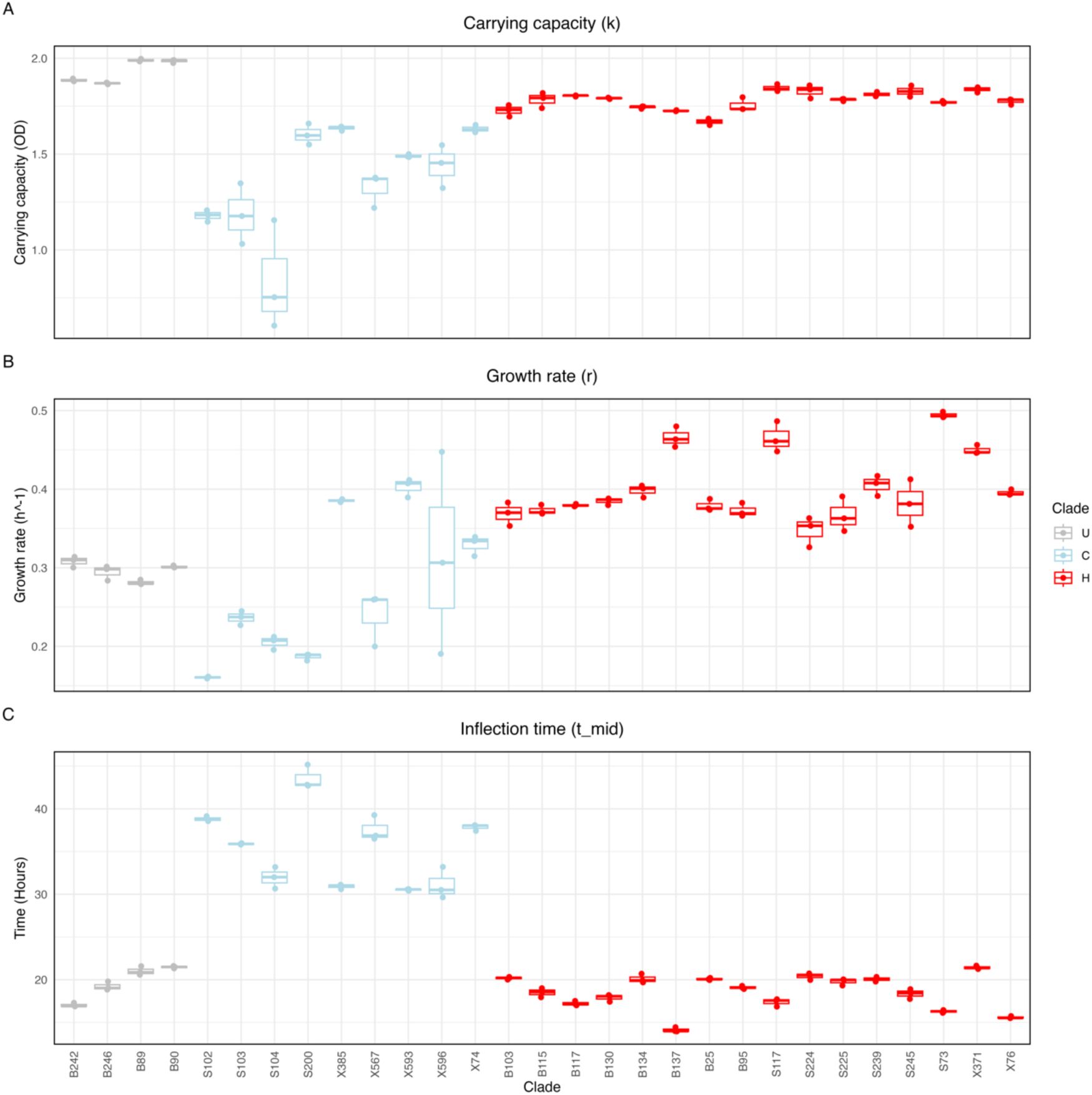
Boxplots depicting the carrying capacity (A), growth rate (B) and inflection time (C) of each isolate.
The boxplots were coloured by clade. Measurements were obtained from three independent technical replicates for each isolate.
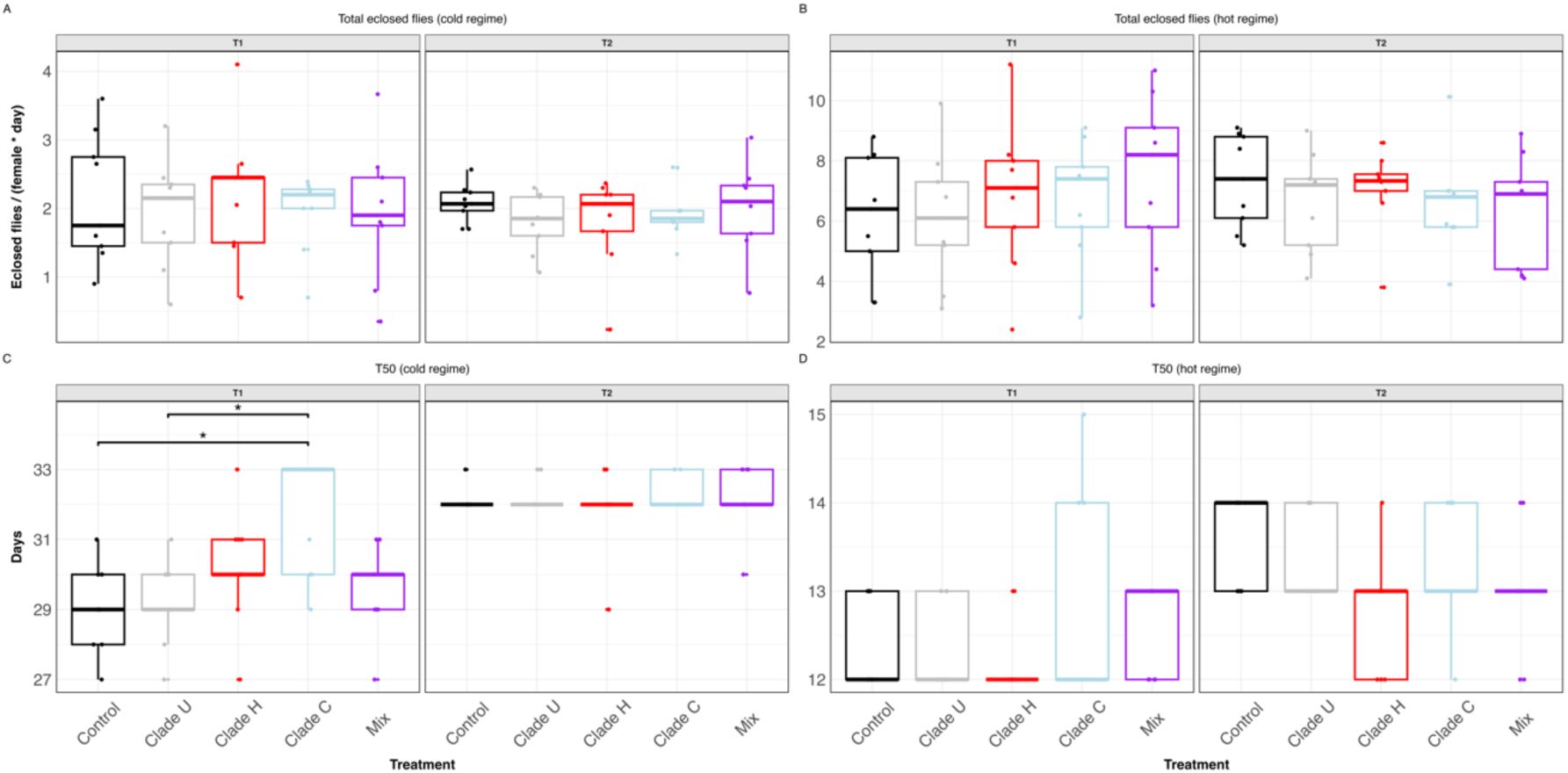
Fitness effect of
L. plantarum
inoculation in conventionally reared
D. simulans
.
(A, B) Total number of F1 flies eclosed normalized by day and female under the cold (A) and hot (B) regime. (C, D) Developmental time, estimated as the number of days it takes 50% of the offspring to eclose. Measurements were grouped by inoculation treatment and transfer. Each dot corresponds to a biological replicate (n = 10) Statistical significance was determined using Dunn’s test with Holm-adjusted p-values. Only significant comparisons are indicated. **** p < 0.0001; *** p < 0.001; ** p < 0.01; * p < 0.05.
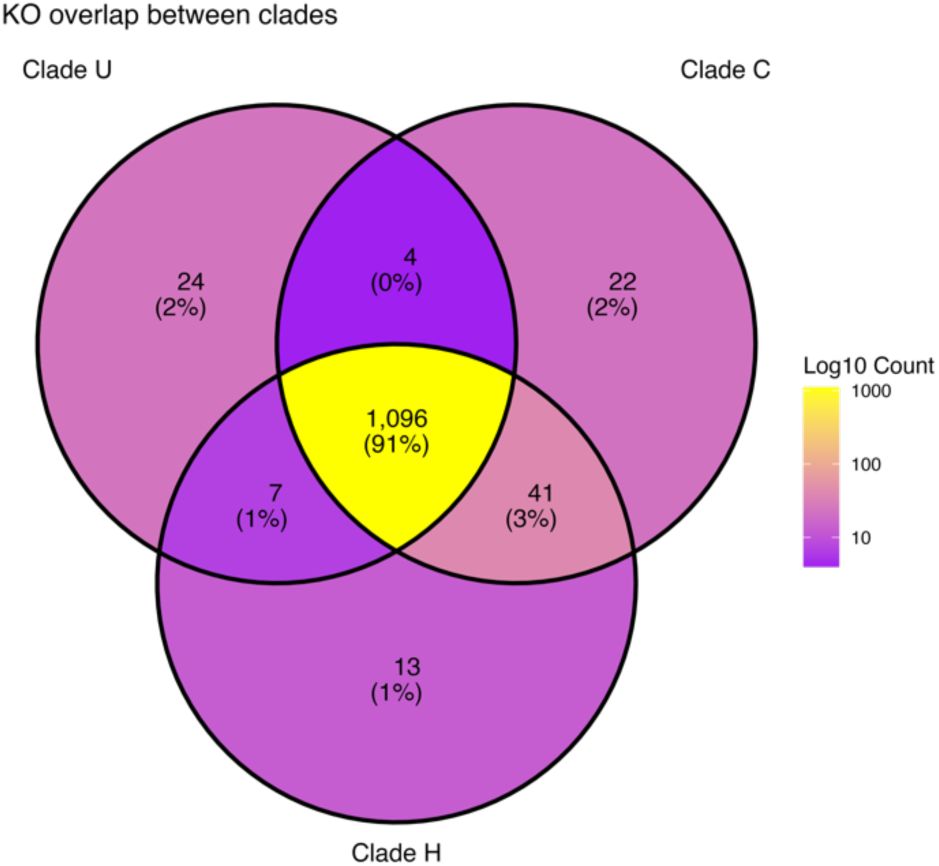
Venn diagram depicting the overlap in KEGG Orthologs between the three clades.
Segments were coloured by number of KOs.
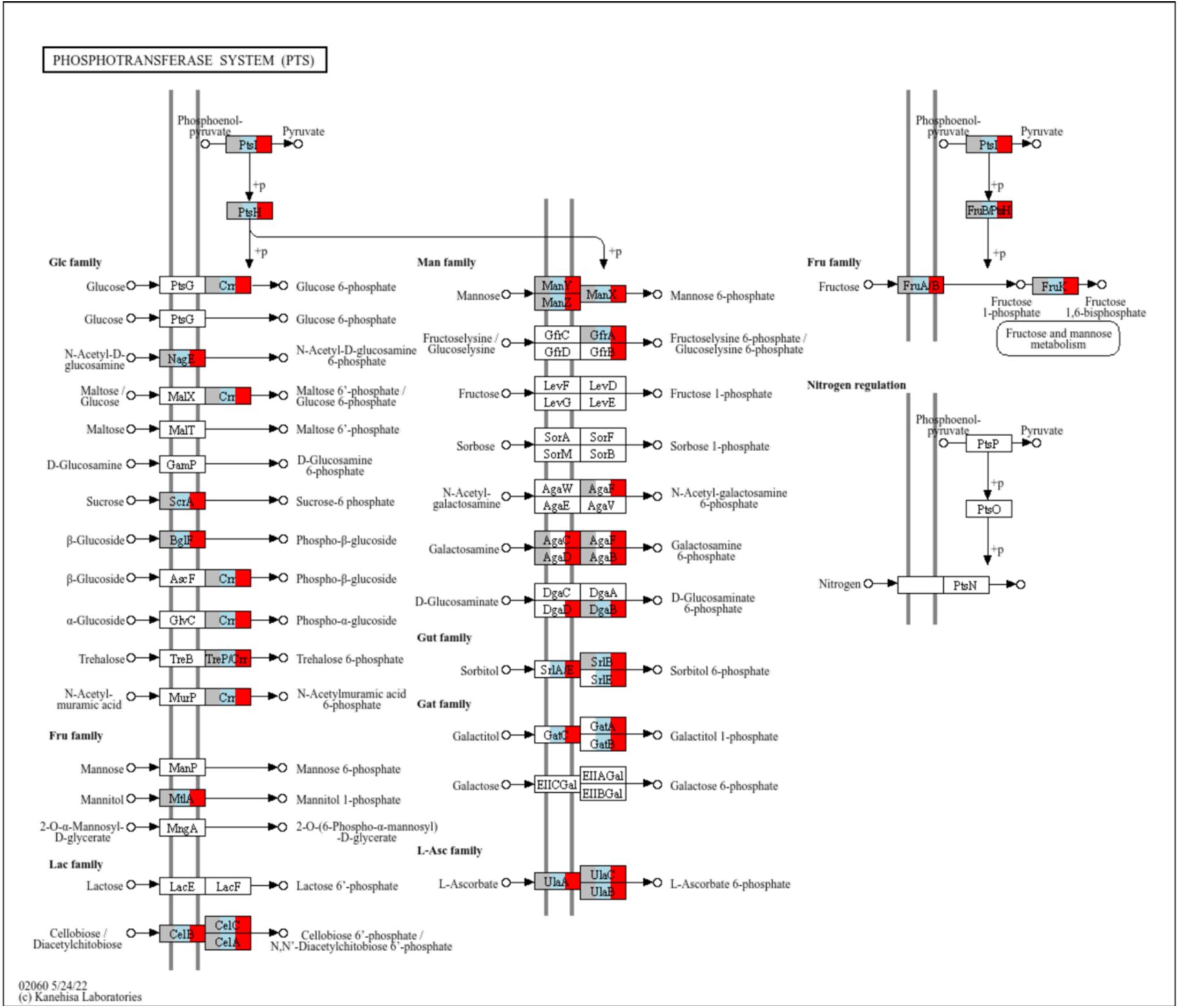
Graphical representation of the KEGG pathway map02060 (Phosphotransferase system).
Each rectangle represents a KEGG Ortholog (KO). Orthologs that are enriched in each clade are coloured in grey (present in clade U), blue (present in clade C) and/or red (present in clade H). The three clades differ in the set of sugar-related PTS transporters. Clades C and H have the capacity to internalize sorbitol and galactitol, whereas clades H and U have the ability to import galactosamine.
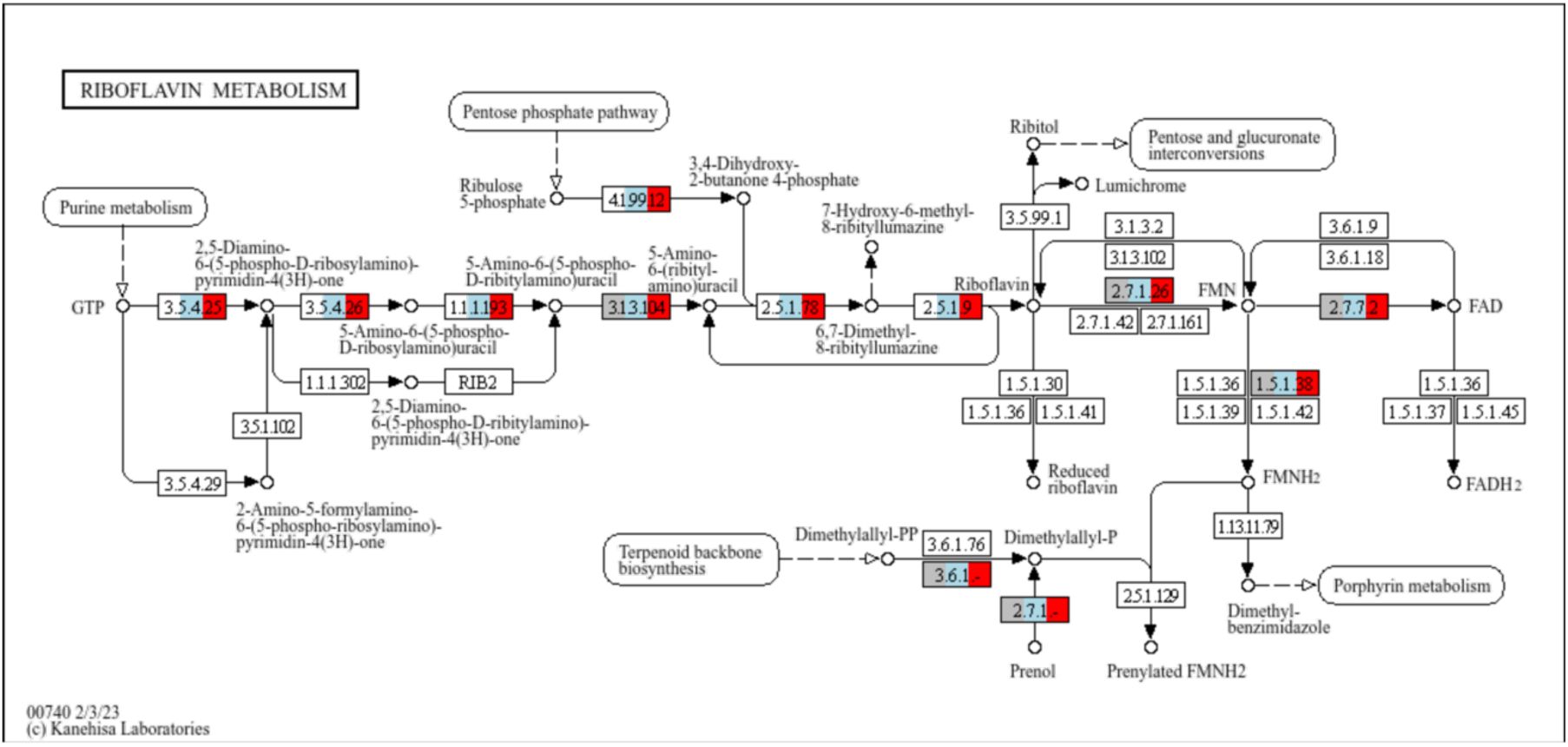
Graphical representation of the KEGG pathway map00740 (Riboflavin metabolism).
Each rectangle represents a KEGG Ortholog (KO). Orthologs that are enriched in each clade are coloured in grey (present in clade U), blue (present in clade C) and/or red (present in clade H). Clade U lacks the capacity to produce de novo FAD from GTP, since it is lacking several genes from the pathway.
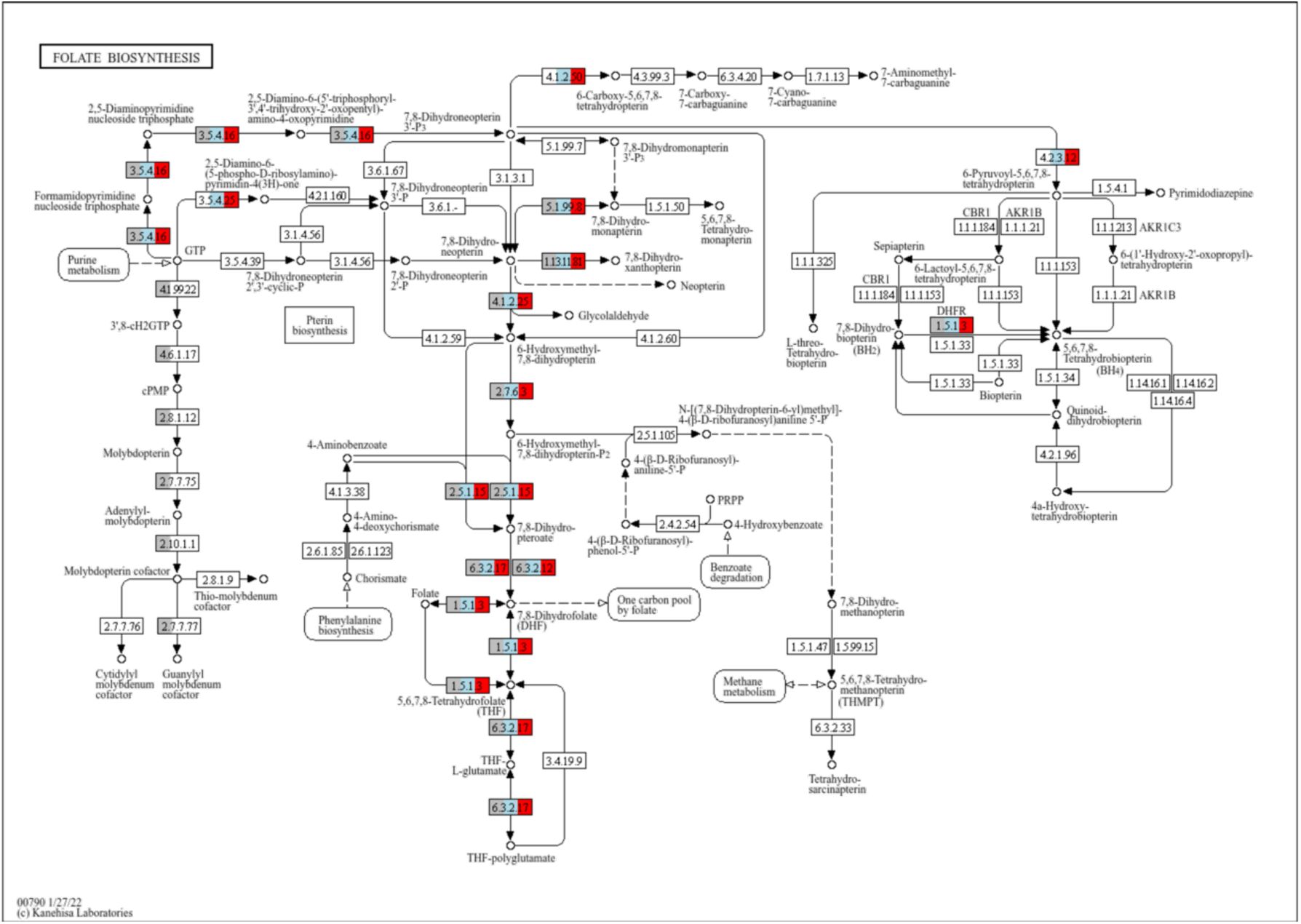
Graphical representation of the KEGG pathway map00790 (Folate biosynthesis).
Each rectangle represents a KEGG Ortholog (KO). Orthologs that are enriched in each clade are coloured in grey (present in clade U), blue (present in clade C) and/or red (present in clade H). Clade U has the unique capacity to synthesize de novo guanylyl molybdenum cofactor.
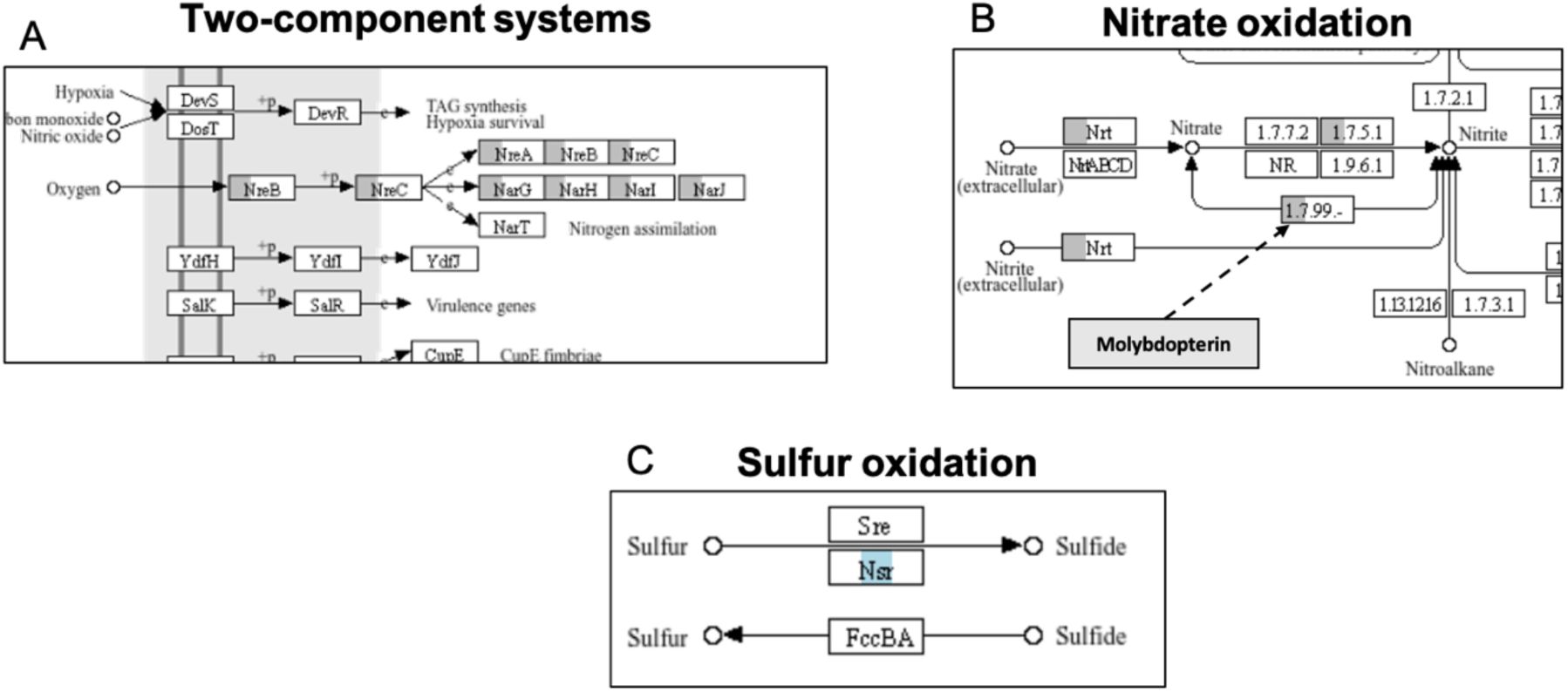
Subset of the KEGG pathways map02020 (Two-component system; A), map00910 (Nitrogen metabolism; B), and map00920 (Sulfur metabolism; C).
Each rectangle represents a KEGG Ortholog (KO). Orthologs that are enriched in each clade are coloured in grey (present in clade U), blue (present in clade C) and/or red (present in clade H). Clade U encodes a unique alternative respiratory system. The operons nreABC and narGHIJ encode for genes that sense anoxic conditions and use nitrate as terminal electron donor instead of oxygen (A, B). The enzyme sulfur oxidoreductase (Nsr) is highly enriched in the clade C, which suggests that it can potentially use sulfur as terminal electron acceptor in absence of oxygen (C).
Data availability
The sequencing raw data used in this study are deposited in the European Nucleotide Archive (ENA) under the BioProject accessions: PRJEB96332, PRJEB29281, PRJEB20780, PRJEB20533, and PRJEB15225 (See
supplementary tables S1 and S4
). All codes and material needed for data processing and reproduction can be found in
.
Acknowledgements
We thank Cameron Strachan and Evelyne Selberherr from the Zentrum für Lebensmittelmikrobiologie (Veterinärmedizinische Universität Wien) for granting us access to a plate reader for the growth experiment. We also wish to thank Martin Polz, William Ludington, Cameron Strachan and Xiaoqian Annie Yu for their feedback regarding experimental design and interpretation of the results. Finally, we are grateful to all members of the Institut für Populationsgenetik (Veterinärmedizinische Universität Wien) for helpful discussions and feedback throughout the project.
Additional information
Author Contributions
Bosco Gracia-Alvira: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Software, Visualization, Writing – original draft. Stefanie Migotti: Resources, Writing – review and editing. Xiaomeng Tian: Resources, Writing – review and editing. Viola Nolte: Data curation, Resources. Christian Schlötterer: Conceptualization, Project administration, Funding acquisition, Resources, Supervision, Writing – review and editing.
Funding
Austrian Science Fund (FWF)
Christian Schlötterer
Austrian Science Fund (FWF)
Christian Schlötterer
Austrian Science Fund (FWF)
Christian Schlötterer
EC | European Research Council (ERC) (Achadapt)
Christian Schlötterer
Additional files
References
1.
Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences
Nat Biotechnol
31
:814–21
PubMed
Google Scholar
2.
PICRUSt2 for prediction of metagenome functions
Nat Biotechnol
38
:685–8
PubMed
Google Scholar
3.
Assessing the Unseen Bacterial Diversity in Microbial Communities
Genome Biol Evol
7
:3416–25
PubMed
Google Scholar
4.
Diversity within species: interpreting strains in microbiomes
Nat Rev Microbiol
18
:491–506
PubMed
Google Scholar
5.
Diversification of Escherichia coli genomes: are bacteriophages the major contributors?
Trends Microbiol
9
:481–5
https://doi.org/10.1016/s0966-842x(01)02173-4
PubMed
Google Scholar
6.
Functional diversity enables multiple symbiont strains to coexist in deep-sea mussels
Nat Microbiol
4
:2487–97
PubMed
Google Scholar
7.
Strain-level diversity of symbiont communities between individuals and populations of a bioluminescent fish
ISME J
17
:2362–9
PubMed
Google Scholar
8.
Impact of intraspecific variation in insect microbiomes on host phenotype and evolution
ISME J
17
:1798–807
PubMed
Google Scholar
9.
Strain competition restricts colonization of an enteric pathogen and prevents colitis
EMBO Rep
17
:1281–91
PubMed
Google Scholar
10.
Ecological Stability Emerges at the Level of Strains in the Human Gut Microbiome
mBio
14
:e0250222
PubMed
Google Scholar
11.
Gut microbiota strain richness is species specific and affects engraftment
Nature
637
:422–9
PubMed
Google Scholar
12.
Single-Cell Genomics Reveals Hundreds of Coexisting Subpopulations in Wild Prochlorococcus
Science
344
:416–20
PubMed
Google Scholar
13.
Microdiversity ensures the maintenance of functional microbial communities under changing environmental conditions
ISME J
13
:2969–83
PubMed
Google Scholar
14.
Quantifying the changes in genetic diversity within sequence-discrete bacterial populations across a spatial and temporal riverine gradient
ISME J
13
:767–79
PubMed
Google Scholar
15.
Nomadic lifestyle of Lactobacillus plantarum revealed by comparative genomics of 54 strains isolated from different habitats
Environ Microbiol
18
:4974–89
PubMed
Google Scholar
16.
Host-Intestinal Microbiota Mutualism: “Learning on the Fly”
Cell Host Microbe
13
:8–14
PubMed
Google Scholar
17.
Bacterial Adaptation to the Host’s Diet Is a Key Evolutionary Force Shaping Drosophila-Lactobacillus Symbiosis
Cell Host Microbe
24
:109–119
PubMed
Google Scholar
18.
Drosophila Perpetuates Nutritional Mutualism by Promoting the Fitness of Its Intestinal Symbiont Lactobacillus plantarum
Cell Metab
27
:362–377
PubMed
Google Scholar
19.
Taxon-Specific Effects of Lactobacillus on Drosophila Host Development
Microb Ecol
79
:241–51
PubMed
Google Scholar
20.
An ANI gap within bacterial species that advances the definitions of intra-species units
mBio
15
:e02696–23
PubMed
Google Scholar
21.
Probabilistic Invasion Underlies Natural Gut Microbiome Stability
Curr Biol
27
:1999–2006
PubMed
Google Scholar
22.
Microdiversity shapes the traits, niche space, and biogeography of microbial taxa
Environ Microbiol Rep
9
:55–70
PubMed
Google Scholar
23.
How do microbes grow in nature? The role of population dynamics in microbial ecology and evolution
EcoEvoRxiv
Google Scholar
24.
Lactobacillus plantarum Promotes Drosophila Systemic Growth by Modulating Hormonal Signals through TOR-Dependent Nutrient Sensing
Cell Metab
14
:403–14
PubMed
Google Scholar
25.
Lactobacillus plantarum favors the early emergence of fit and fertile adult Drosophila upon chronic undernutrition
J Exp Biol
220
:900–7
PubMed
Google Scholar
26.
Pathogen Virulence Impedes Mutualist-Mediated Enhancement of Host Juvenile Growth via Inhibition of Protein Digestion
Cell Host Microbe
18
:445–55
PubMed
Google Scholar
27.
Simple animal models for microbiome research
Nat Rev Microbiol
17
:764–75
PubMed
Google Scholar
28.
Stochastic microbiome assembly depends on context
Proc Natl Acad Sci
119
:e2115877119
PubMed
Google Scholar
29.
Higher-order microbiome interactions and how to find them
Trends Microbiol
PubMed
Google Scholar
30.
Monoassociation with Lactobacillus plantarum Disrupts Intestinal Homeostasis in Adult Drosophila melanogaster
mBio
9
PubMed
Google Scholar
31.
Bacterial chitin degradation—mechanisms and ecophysiological strategies
Front Microbiol
4
PubMed
Google Scholar
32.
Microbiome composition shapes rapid genomic adaptation of Drosophila melanogaster
Proc Natl Acad Sci
116
:20025–32
PubMed
Google Scholar
33.
Microbial shifts associated to ENSO-derived thermal anomalies reveal coral acclimation at holobiont level
Sci Rep
13
:22049
PubMed
Google Scholar
34.
Host and microbiome jointly contribute to environmental adaptation
ISME J
17
:1953–65
PubMed
Google Scholar
35.
Gut microbiota facilitate adaptation of invasive moths to new host plants
ISME J
18
:wrae031
PubMed
Google Scholar
36.
Low-diversity bacterial community in the gut of the fruitfly Drosophila melanogaster
Environ Microbiol
13
:1889–900
PubMed
Google Scholar
37.
Drosophila as a model for the gut microbiome
PLOS Pathog
16
:e1008398
PubMed
Google Scholar
38.
Lactobacilli-Host mutualism: ‘learning on the fly’
Microb Cell Factories
13
:S6
PubMed
Google Scholar
39.
One species, many faces: The underappreciated importance of strain diversity
PLOS Pathog
20
:e1011931
PubMed
Google Scholar
40.
A genomic investigation of ecological differentiation between free-living and Drosophila-associated bacteria
Mol Ecol
26
:4536–50
PubMed
Google Scholar
41.
Strong responses of Drosophila melanogaster microbiota to developmental temperature
Fly
12
:1–12
PubMed
Google Scholar
42.
Experimental temperatures shape host microbiome diversity and composition
Glob Change Biol
29
:41–56
PubMed
Google Scholar
43.
Temperature Adaptation of Aquatic Bacterial Community Growth Is Faster in Response to Rising than to Falling Temperature
Microb Ecol
87
:38
PubMed
Google Scholar
44.
Drosophila simulans: A Species with Improved Resolution in Evolve and Resequence Studies
G3
7
:2337–43
PubMed
Google Scholar
45.
Evolution of phenotypic variance in response to a novel hot environment
Mol Ecol
31
:934–45
PubMed
Google Scholar
46.
Sequencing pools of individuals — mining genome-wide polymorphism data without big funding
Nat Rev Genet
15
:749–63
PubMed
Google Scholar
47.
Genetic redundancy fuels polygenic adaptation in Drosophila
PLOS Biol
17
:e3000128
PubMed
Google Scholar
48.
Experimental evolution reveals habitat-specific fitness dynamics among W olbachia clades in D rosophila melanogaster
Mol Ecol
23
:802–14
PubMed
Google Scholar
49.
Long-Term Dynamics Among Wolbachia Strains During Thermal Adaptation of Their Drosophila melanogaster Hosts
Front Genet
11
:482
PubMed
Google Scholar
50.
Long-term gut microbiome dynamics in Drosophila melanogaster reveal environment-specific associations between bacterial taxa at the family level
Proc R Soc B Biol Sci
288
:20212193
PubMed
Google Scholar
51.
A simple genetic basis of adaptation to a novel thermal environment results in complex metabolic rewiring in Drosophila
Genome Biol
19
:119
PubMed
Google Scholar
52.
The genetic architecture of temperature adaptation is shaped by population ancestry and not by selection regime
Genome Biol
22
:211
PubMed
Google Scholar
53.
A simple salting out procedure for extracting DNA from human nucleated cells
Nucleic Acids Res
16
:1215
PubMed
Google Scholar
54.
An Efficient DNA Extraction Method for Lactobacillus casei, a Difficult-to-Lyse Bacterium
Int J Enteric Pathog
4
:e32472
Google Scholar
55.
BBMap
SourceForge
56.
Using SPAdes De Novo Assembler
Curr Protoc Bioinforma
70
:e102
PubMed
Google Scholar
57.
CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning
Nat Methods
20
:1203–12
PubMed
Google Scholar
58.
GTDB-Tk v2: memory friendly classification with the genome taxonomy database
Bioinformatics
38
:5315–6
PubMed
Google Scholar
59.
Centrifuge: rapid and sensitive classification of metagenomic sequences
Genome Res
PubMed
Google Scholar
60.
Prokka: rapid prokaryotic genome annotation
Bioinforma Oxf Engl
30
:2068–9
PubMed
Google Scholar
61.
Roary: rapid large-scale prokaryote pan genome analysis
Bioinformatics
31
:3691–3
PubMed
Google Scholar
62.
SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments
Microb Genomics
2
:e000056
PubMed
Google Scholar
63.
IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era
Mol Biol Evol
37
:1530–4
PubMed
Google Scholar
64.
Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens
Anal Methods
8
:12–24
Google Scholar
65.
Anvi’o: an advanced analysis and visualization platform for ‘omics data
PeerJ
3
:e1319
PubMed
Google Scholar
66.
Linking pangenomes and metagenomes: the Prochlorococcus metapangenome
PeerJ
6
:e4320
PubMed
Google Scholar
67.
Functional and genetic markers of niche partitioning among enigmatic members of the human oral microbiome
Genome Biol
21
:292
PubMed
Google Scholar
68.
Imoto S. ggkegg: analysis and visualization of KEGG data utilizing the grammar of graphics
Bioinformatics
39
:btad622
PubMed
Google Scholar
69.
Fast gapped-read alignment with Bowtie 2
Nat Methods
9
:357–9
PubMed
Google Scholar
70.
Exploring environmental intra-species diversity through non-redundant pangenome assemblies
Mol Ecol Resour
PubMed
Google Scholar
71.
Simulating Illumina metagenomic data with InSilicoSeq
Bioinformatics
35
:521–2
PubMed
Google Scholar
72.
SeqKit2: A Swiss army knife for sequence and alignment processing
iMeta
3
:e191
PubMed
Google Scholar
73.
Growthcurver: an R package for obtaining interpretable metrics from microbial growth curves
BMC Bioinformatics
17
:172
PubMed
Google Scholar
74.
R: A Language and Environment for Statistical Computing
Vienna, Austria
:
R Foundation for Statistical Computing
75.
rstatix: Pipe-Friendly Framework for Basic Statistical Tests
76.
Generating Germ-Free Drosophila to Study Gut-Microbe Interactions: Protocol to Rear Drosophila Under Axenic Conditions
Curr Protoc Toxicol
77
:e52
PubMed
Google Scholar
77.
Transaldolase: From biochemistry to human disease
Int J Biochem Cell Biol
41
:1482–94
PubMed
Google Scholar
78.
Bacterial chitin degradation—mechanisms and ecophysiological strategies
Front Microbiol
4
PubMed
Google Scholar
79.
Overlapping riboflavin supply pathways in bacteria
Crit Rev Microbiol
43
:196–209
PubMed
Google Scholar
80.
Molybdenum cofactors, enzymes and pathways
Nature
460
:839–47
PubMed
Google Scholar
81.
Prokaryotic Nitrate Reduction: Molecular Properties and Functional Distinction among Bacterial Nitrate Reductases
J Bacteriol
181
:6573–84
PubMed
Google Scholar
82.
The Nitrate Reductase and Nitrite Reductase Operons and the narT Gene of Staphylococcus carnosus Are Positively Controlled by the Novel Two-Component System NreBC
J Bacteriol
184
:6624–34
PubMed
Google Scholar
83.
Role of narK2X and narGHJI in Hypoxic Upregulation of Nitrate Reduction by Mycobacterium tuberculosis
J Bacteriol
185
:7247–56
PubMed
Google Scholar
84.
Synthesis of L-2,3-Diaminopropionic Acid, a Siderophore and Antibiotic Precursor
Chem Biol
21
:379–88
PubMed
Google Scholar
85.
Inhibition of enzymic transamination of aspartic acid by hydroxyaspartate, 2,3-diaminosuccinate and 2,3-diaminopropionate
Biochim Biophys Acta
24
:78–82
https://doi.org/10.1016/0006-3002(57)90148-8
PubMed
Google Scholar
86.
Functional Analysis of the Genes Encoding Diaminopropionate Ammonia Lyase in Escherichia coli and Salmonella enterica Serovar Typhimurium
J Bacteriol
194
:5604–12
PubMed
Google Scholar
Intraspecific diversity of Lactiplantibacillus plantarum reflects functional diversification in the microbiome of Drosophila simulans
ENA
ID PRJEB96332
Genetic redundancy fuels polygenic adaptation in Drosophila
ENA
ID PRJEB29281
Drosophila simulans: a species with improved resolution in Evolve and Resequence studies
ENA
ID PRJEB20780
P-element invasion in Drosophila simulans
ENA
ID PRJEB20533
Ancestral population reconstitution from isofemale lines as a tool for experimental evolution
ENA
ID PRJEB15225
Article and author information
Author information
Cite all versions
You can cite all versions using the DOI
10.7554/eLife.110808
. This DOI represents all versions, and will always resolve to the latest one.
Copyright
© 2026,
Gracia-Alvira et al.
This article is distributed under the terms of the
Creative Commons Attribution License
, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
views
0
downloads
0
citations
0
Views, downloads and citations are aggregated across all versions of this paper published by eLife.





