 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库周末文摘 | 基于监管要求的临床试验元数据审核实操指南

引用本文



颜崇超.基于监管要求的临床试验元数据审核实操指南[J].中国食品药品监管.2026.04(267):104-123.
基于监管要求的临床试验元数据审核实操指南
A Practical Guide to Metadata Review in Clinical Trials for Regulatory Compliance
颜崇超
江苏恒瑞医药生物计量部临床数据科学中心
YAN Charles
Clinical Data Science Center, Biometrics Department, Jiangsu Hengrui Pharmaceuticals Co., Ltd.
摘 要Abstract
随着全球临床试验监管体系持续完善,元数据作为保障数据全生命周期可追溯、可验证的核心载体,其审核质量直接影响试验数据可靠性与监管认可度。针对当前行业存在的元数据定义不统一、盲态保护不完善、第三方管理薄弱、技术工具适配不足等问题,本研究参考《E6(R3):药物临床试验质量管理规范技术指导原则》《药物临床试验计算机化系统和电子数据指导原则(征求意见稿)》等国内外相关要求,结合临床实操经验,系统构建了兼具合规性与实操性的临床试验元数据审核框架。研究明确了技术元数据、过程元数据、业务元数据的核心范畴与临床价值,确立“ 范围全覆盖、内容全维度、对象分风险、实施全流程” 4 大审核原则,细化了数据采集、处理、传输、存储、归档、销毁全生命周期的审核要点,制定盲态数据的元数据审核专项方案、第三方管理机制及工具选型标准,并针对行业痛点提出标准化体系建设、技术工具升级、人员能力提升等优化对策。该框架为临床试验元数据审核提供了标准化实操参考,可有效提升数据质量与审核效率,降低监管核查风险,助力临床试验数据治理与国际监管要求接轨。
With the continuous evolution of global regulatory requirements for clinical trials, metadata has become a core element for ensuring the traceability and verifiability of data throughout its lifecycle. The quality of metadata review directly affects the reliability of trial data and its acceptance by regulatory authorities. In response to common industry challenges, such as inconsistent metadata definitions, inadequate blinding protection, weak third-party management, and insufficient adaptation of technical tools, this study systematically constructs a compliant and practice-oriented metadata review framework for clinical trials. The framework is grounded in both international and domestic regulatory requirements, including ICH GCP E6 (R3) and China’s Guidelines for Computerized Systems and Electronic Data in Clinical Trials (Draft) , and is informed by real-world operational experience. The study clarifies the core scope and clinical value of technical, process, and business metadata, and establishes four key review principles: full-scope coverage, multidimensional content, risk-based object classification, and lifecycle-wide implementation. It further elaborates review focus areas across the entire data lifecycle (collection, processing, transformation, storage, archiving, and destruction). In addition, it proposes dedicated metadata review plan for blinded data, third-party management, and tool selection, and puts forward optimization strategies targeting key industry challenges, such as standardized system development, technological enhancement, and workforce capacity building. This framework provides a standardized and practical reference for metadata review in clinical trials, contributing to improved data quality and review efficiency, reduced regulatory inspection risks, and better alignment of clinical trial data governance with international regulatory expectations.
关键词Key words
元数据审核;数据完整性;盲态保护;数据全生命周期;监管合规
metadata review; data integrity; blinding; data lifecycle; regulatory compliance

临床试验数据是评价药物有效性与安全性的核心依据,其质量直接关系药品上市审批决策的科学性与公众用药安全。近年来,全球临床试验监管体系持续完善,数据全生命周期治理已成为行业共识,国内外药品监管机构,特别是国家药品监督管理局药品审评中心发布的《药物临床试验计算机化系统和电子数据指导原则(征求意见稿)》,对电子数据的可追溯性、完整性、准确性提出更为严格的要求[1-3]。元数据作为描述数据属性、背景及处理过程的“数据字典”,贯穿临床试验数据采集、处理、传输、存储、归档与销毁全生命周期,是保障数据全链条可追溯、可验证的核心技术载体。元数据审核的质量也直接影响试验结果的可信度与监管认可度[4-5]。
当前,临床试验行业在元数据管理实践中仍面临诸多挑战,包括:元数据定义与范畴不统一导致审核范围模糊、盲态保护措施不完善引发数据泄露风险、第三方协作过程中责任边界不清造成审核遗漏、技术工具适配不足影响审核效率与准确性等[6]。在实际操作中,申办者通常未建立元数据审核的标准化操作规程,临床试验的元数据管理问题在监管核查中常被要求做额外的补充说明[7]。因此,构建一套符合国内外监管要求、兼具实操性与系统性的元数据审核框架,对规范临床试验数据治理流程、提升数据质量、降低监管风险具有重要现实意义。本文基于临床数据治理的实操经验与监管实践总结,系统梳理了元数据审核的核心要点与实施路径,可为行业提供标准化与专业化的参考。
1 临床试验元数据的定义、范畴与核心临床价值
1.1 元数据的定义与范畴
临床试验元数据是描述电子数据“ 背景信息”“流转轨迹” 及“业务逻辑” 的关键载体,具体指定义数据属性、记录处理过程、说明数据关联关系的各类结构化与非结构化信息,与源数据(即原始数据)共同构成临床试验数据的完整体系[8-11]。在临床实践中,元数据与源数据是既相互关联又明确区分的核心概念,二者的核心差异可通过表1 清晰界定。

在实际操作中,需明确区分两类核心元数据[12-14] :一类是试验级元数据,与特定临床试验直接相关, 如参与者疗效指标的产生与修改轨迹、实验室数据传输映射关系等, 一般由临床数据管理员主导审核。另一类是系统级元数据,支撑临床试验计算机化系统[ 如电子数据采集(electronic data capture,EDC)系统] 运行的基础信息,包括服务器配置日志、数据库备份记录等,由系统管理部门或技术支持部门负责审核、验证与管控。
结合临床数据治理实操场景,元数据可细分为3 类核心类型,各类型具体构成情况如下。
1.1.1 技术元数据
聚焦数据采集与存储的技术环境信息,是保障数据采集准确性与存储安全性的基础,主要包括:①数据采集设备相关信息。如设备唯一标识符(指用于唯一识别单台物理设备的资产编号或序列号,用于设备管理与溯源)、型号版本、固件编号、最后校准日期、校准证书编号、校准机构资质、设备故障记录、维修日志、设备使用人员授权记录等,以确保采集设备处于合规可用状态。②计算机化系统配置信息。如系统版本号、数据库类型、服务器 IP 地址、接口通信协议、系统验证状态、系统补丁更新记录等,以保障系统运行的稳定性与数据处理的规范性。③存储与备份相关信息。如数据存储物理路径、云存储账号权限、备份频率、备份介质、加密算法、归档格式、恢复测试记录等,以确保数据长期安全存储与快速恢复。
1.1.2 过程元数据
记录数据全流程操作轨迹,是实现数据可追溯性的核心,主要包括:①稽查轨迹。如数据原始值、修改后新值、操作人账号、操作时间戳[ 统一采用协调世界时(universal time coordinated,UTC)标准,精确至秒]、修改原因、审批人账号、审批时间等,以完整呈现数据变更全流程。②访问日志。如用户登录/ 退出时间、访问模块、操作类型(查询、修改、删除)、权限变更记录、登录IP 地址、终端设备信息等,以监控数据访问行为的合规性。③工作流程相关信息。如数据录入时间、逻辑核查触发记录、质疑创建/ 回复/ 关闭记录、数据更正审批流程、数据冻结与锁定/ 解冻与解锁的申请与审批、操作的记录,数据签字确认的记录以及数据清理确认、源数据核查确认以及医学审核确认的记录等,以规范数据处理流程。④事件日志。如系统故障发生时间、影响范围、故障原因分析、处理措施、恢复时间,数据传输异常记录、网络安全事件记录等,以保障试验流程连续性。
1.1.3 业务元数据
界定数据的业务逻辑与标准,是保障数据一致性与可理解性的关键,主要包括:①数据点定义。如变量名称、数据类型、取值范围、变量编码规则(如性别数据库变量编码:M= 男,F= 女)、临床意义说明等,以确保数据采集的标准与统一。②逻辑核查规则。如核查条件、触发阈值、异常提示信息、规则生效时间、规则变更记录等,以保障数据采集的质量。③衍生数据计算方法。如计算公式、参数设置、溯源路径、统计软件版本等,以确保衍生数据可重复验证。④数据核查(如源数据核查、数据清理、医学审核)。⑤研究者签名以及数据点的状态(草稿、提交、审核中、冻结、锁定以及解冻或解锁等)。⑥医学编码标准。如医学编码词典版本、编码映射关系、编码人员资质证书编号、编码复核记录等,以保障数据编码准确性。
在临床试验数据治理实践中,需明确关键元数据清单,将系统验证、稽查轨迹、设备校准记录、权限变更日志、随机化与盲态维护相关信息、衍生数据计算逻辑等纳入强制审核范围,确保无核心元数据缺失[15-16]。
1.2 元数据的核心临床价值
1.2.1 筑牢数据可靠性基础,保障临床试验结果科学可信
元数据是实现临床试验数据可靠性核心原则的技术支撑,通过完整记录临床试验数据全生命周期信息,确保数据可归因、易读、同时、原始、准确、完整、一致、持久、可获得且可追溯 [17]。例如,某Ⅱ期抗肿瘤药物临床试验中,通过稽查轨迹发现 2 例关键疗效指标(客观缓解率)修改未记录审批流程,经元数据审核及时补充审批记录与原始佐证材料,避免了因数据可靠性缺陷导致监管核查的问题。某糖尿病药物临床试验中,通过设备元数据发现 1台血糖检测仪未按要求校准,涉及 5 例参与者数据,及时发现后,使用经校准的血糖检测仪,重新采集数据,保障了数据准确性。
1.2.2 支撑跨境监管合规,降低核查风险
当前全球主要药品监管机构均将元数据审核作为临床试验核查的核心内容,标准化的元数据审核可同时满足不同地区监管要求,减少跨境试验的合规成本[7,16,18]。例如,某国际多中心药物临床试验中,通过访问日志、权限变更记录、操作轨迹元数据,清晰呈现非盲权限的授权与使用流程,最终顺利通过国内与国际药品监管机构的核查。另一跨境临床试验因未完整保留数据迁移元数据(含数据传输日志、接口日志、校验记录、版本轨迹、迁移时间戳、数据映射关系、完整性校验值),导致监管机构无法验证数据传输完整性,被要求额外补充数据完整性的证据,延长了申报周期。
1.2.3 精准识别数据异常风险,强化质量管控
元数据审核是发现数据造假与系统性问题的有效手段[19-20]。例如,某心血管药物多中心临床试验中, 通过对电子化患者报告结局(electronic patient reported outcome,ePRO) 数据的时间戳、操作账号、IP 地址、录入轨迹、日志记录等元数据审核,发现某临床试验研究中心的参与者日记在01:00~03:00 集中录入,且操作 IP 地址一致,最终证实该数据由临床研究协调员(clinical research coordinator,CRC)代录。通过对上述元数据审核,及时发现并纠正了该临床试验研究中心数据录入不合规问题,避免影响试验结论。在某项Ⅲ期临床试验中,数据管理团队审核逻辑核查规则的版本变更、修改记录、操作人、修改时间、失效原因等元数据时,发现一条安全性相关核查规则被失效。团队立即恢复该规则,及时避免了安全性风险漏查,防范了合规风险。
1.2.4 优化试验流程管理,提升运营效率
通过元数据可视化分析可精准识别临床试验流程瓶颈。例如,某肿瘤药物临床试验中,通过 EDC 系统访问日志分析,发现某临床试验研究中心主要研究者平均审核签字电子病例报告表(electronic case report form,eCRF)时间远超方案规定的时间,立刻启动针对性培训与流程优化,有效缩短了签字周期。某内分泌药物临床试验中,通过设备元数据统计,优化设备调度方案,将样本采集等待时间从 2 h 缩短至30 min。通过统计数据质疑记录,可系统分析由逻辑核查规则触发的异常。数据质疑元数据包括:质疑编号、质疑类型、质疑内容、触发字段、触发时间、处理人、处理时间、处理状态、关闭原因、关联核查规则编号及版本信息等。在某项临床试验中,通过统计上述元数据发现,实验室正常值相关逻辑核查规则触发频率占总质疑数的 20%,经分析后优化核查条件,完善实验室正常值获取与录入流程,显著降低质疑数量与处理工作量。
1.2.5 规范第三方协同管理,明确责任边界
多中心临床试验中,申办者通常委托合同研究组织(contract research organization,CRO)、现场管理组织(site management organization,SMO)、中心实验室以及系统供应商等第三方参与数据治理,元数据可有效界定各方责任。例如,某生物等效性临床试验中,通过中心实验室信息管理系统(laboratory information management system,LIMS)的校准记录与数据传输稽查轨迹元数据,确认实验室数据采集与传输合规性。某疫苗临床试验中,根据项目的要求,通过权限稽查轨迹元数据明确SMO人员仅具备数据录入与质疑回复权限,无数据删除权限,避免因权限滥用导致的数据风险。某多中心临床试验中,通过数据传输稽查轨迹元数据的完整性校验[如比对信息摘要算法第五版(message digest algorithm5,MD5)值]比对结果,界定数据传输的完整性与责任方。MD5作为通用密码散列算法,可对电子数据生成唯一固定长度的数字摘要,用于验证数据在传输、存储过程中未发生篡改、丢失或替换,是保障临床试验数据完整性的核心技术手段。
2 元数据审核的核心要点
元数据审核应遵循“ 范围全覆盖、内容全维度、对象分风险、实施全流程” 4 大核心原则,结合数据全生命周期管理与临床实际操作场景,本文明确了以下核心审核要点。
2.1 审核范围:覆盖数据全生命周期与关键场景
元数据审核需贯穿临床试验数据采集、处理、传输/迁移、存储/归档、系统运维、销毁的完整生命周期,实现全流程无死角覆盖。
2.1.1 数据全生命周期无死角覆盖
(1)数据采集阶段。重点审核设备校准记录有效性,包括设备授权使用记录、录入人员权限与资质的匹配性,时间戳的统一性,以及自动采集数据与手动录入数据的一致性相关的元数据。
(2)数据处理阶段。核心审核稽查轨迹完整性,包括数据更正审批流程、衍生数据计算参数准确性、逻辑核查规则与数据核查执行记录等元数据。
(3) 数据传输/ 迁移阶段。重点审核传输协议合规性,包括数据映射关系文档、传输完整性校验结果、迁移前后数据一致性验证报告等元数据。
(4) 数据存储/ 归档阶段。审核存储路径安全性,包括加密状态、备份记录完整性,归档格式合规性与可读性,保留期限达标情况等元数据。
(5)系统运维阶段。审核用户账号创建/ 变更/ 注销记录、权限变更审批记录、系统升级与补丁更新记录、系统事件日志处理及时性等元数据。
(6)数据销毁阶段。作为数据全生命周期的最终环节,需围绕“ 合规性、可追溯性、安全性”开展审核,确保销毁过程无数据泄露风险,且全程符合监管要求。
2.1.2 五大核心场景专项审核
(1)系统访问行为审核。已完成相关培训的用户可申请系统使用账户,账户创建需经授权并留存审批材料;长期未登录账号(如超过90 d)需按规程及时清理(如失活);重点监控跨临床试验研究中心访问异常(如某用户同时登录不同地区、不同临床试验研究中心系统且操作时间重叠)、异常IP 登录(非临床试验研究中心所在地IP 访问核心数据模块)、权限与角色不匹配(如CRC 数据录入人员被误授予数据审核或者电子签名的权限)等情况。例如,某国际多中心临床试验中,通过定期访问日志发现 1 名已离职SMO 人员账号未及时注销,仍可访问 eCRF 数据,经紧急处理避免数据泄露风险。
(2)数据变更行为审核。重点关注关键数据(主要疗效指标、严重不良事件相关数据)的修改频率(如某参与者同一核心指标修改≥ 3 次,需专项说明)、修改时间分布(如集中在数据库锁定前24 h 批量修改)、修改原因合理性(如“ 录入错误” 需附原始记录相关元数据佐证);数据更正需遵循“ 先质疑后更正” 流程,衍生数据修改需同步更新计算逻辑相关元数据。例如,某Ⅲ期糖尿病药物临床试验中,审核发现某临床试验研究中心将糖化血红蛋白检测值“7.8%” 修改为“ 6.8%”,但未留存原始记录相关元数据,无修改依据且不可追溯,最终判定该数据无效。
(3)数据采集行为审核。验证采集时间与方案要求的一致性( 如给药后8 h 采集的血药浓度数据,时间戳需在给药时间±15 min 内);核查自动采集设备的样本采集时间与手动录入数据的一致性;关注 ePRO 数据采集合理性(如参与者在非常规时间连续录入多日数据且无修改痕迹)。某 ePRO 临床试验中,通过时间戳与设备元数据核实,发现数例数据由CRC 违规代录,不符合数据采集规范,发现问题后及时完成整改。
(4)报告环节相关审核。审核数据导出时间、导出人员、导出格式、导出筛选条件等元数据;审核报告生成逻辑与业务元数据的一致性(如统计分析报告中“ 有效病例数” 计算逻辑需与衍生数据定义完全匹配);记录报告修订版对应的元数据变更情况。例如,某生物等效性临床试验中,外部样本检测数据与 EDC 系统采集样本存在不一致,通过元数据审核定位原因,即EDC 系统内样本采集信息填写错误,发现问题后及时完成整改。
(5)设备相关审核。核查设备校准记录的连续性(如血糖检测仪需每3 个月校准 1 次);分析设备故障日志与数据采集的关联性(如某离心机在采集样本期间发生故障,需确认故障前后的样本数据是否受到了影响);确保EDC 系统版本及其eCRF 版本与伦理审批方案的一致性(如各临床试验研究中心应使用与伦理审批方案相对应版本的eCRF)[1]。例如,某心血管药物临床试验中,通过设备元数据审核发现 2 台心电图仪校准过期,涉及 8 例参与者数据,经校准后,数据重新采集,保障数据准确性。
2.2 核心审核内容:“五性”验证与风险分层
2.2.1 准确性验证
核查元数据与实际操作的一致性,包括设备校准记录与校准证书信息完全匹配、时间戳与操作实际时间无偏差、数据映射关系与传输方案一致;验证系统自动生成元数据的准确性,如访问日志中的操作类型与实际操作一致、稽查轨迹中的修改原因与质疑记录匹配。
2.2.2 完整性核查
关键元数据无缺失,如稽查轨迹必须包含“ 原始值- 新值-操作人- 时间戳- 修改原因” 5大核心要素,设备元数据需完整记录校准周期、校准结果及校准机构信息;数据处理全流程元数据无断点,指数据产生、质疑、更正、审批、锁定至归档的全过程均应形成连续可追溯的元数据链,任一环节不得缺失或断链,数据更正需与质疑记录、审批记录相互关联,形成完整闭环。例如,某Ⅲ期肿瘤药物临床试验中,研究中心使用EDC 系统录入实验室数据,审核发现约5% 的实验室参考值数据记录缺少操作者ID(仅显示“系统管理员”),原因是该研究中心为图省事,使用管理员账户登录,发现问题后要求其立即整改。
2.2.3 一致性审核
元数据审核应与既定规程一致, 主要包括两方面内容:一是内部规程一致性, 即逻辑核查规则与数据管理计划(data management plan, DMP)或数据审核计划(data validation plan,DVP)相符,衍生数据计算方法与DVP 规定一致、用户权限配置与岗位职责及角色分工匹配。二是跨主体一致性,如多中心临床试验的各中心元数据定义、采集规范与编码标准统一,系统升级、配置变更后元数据标准与变更控制规程保持连贯。例如,某CRO 承担的多中心临床试验中,数据审核团队仅检查原始数据准确性,未按规程开展元数据完整性与合理性审核;某临床研究中心约20% 的“数据修改”记录原因栏填写为“系统错误”,后经核查系统运行正常,该表述与实际情况不符,属于不合规的随意填写。
2.2.4 合规性审核
稽查轨迹功能应处于启用状态,严禁未经授权的禁用或篡改;仅在确需实施隐私保护等特殊情形时方可对相关内容进行处理,且必须完整记录操作行为、处理依据及审批流程,形成可追溯闭环。权限管理应遵循“最小必要”原则,用户访问权限与其岗位职责相匹配,人员调岗或离职后权限应及时注销或失效,所有权限变更均需留存完整审批记录。元数据保存期限应符合相关法规要求,通常需至少保存至药品上市后 5 年或临床试验终止后 10 年(以期限较长者为准);元数据销毁需严格按照既定规程执行,并对销毁过程与结果进行全程记录。
2.2.5 安全性审核
元数据存储与传输加密合规,系统需可提供完整的用户访问与操作的日志记录[ 包括访问日志(access log) 或用户活动日志(user activity log)],无未经授权访问痕迹;备份与灾备合规,备份频率符合相关规程,备份数据可恢复,灾难恢复计划需经测试验证。
2.2.6 风险分层审核方法
本文结合数据重要性、系统复杂度、操作风险、人员资质4个维度建立了风险评估矩阵,以实施差异化审核,见表2。
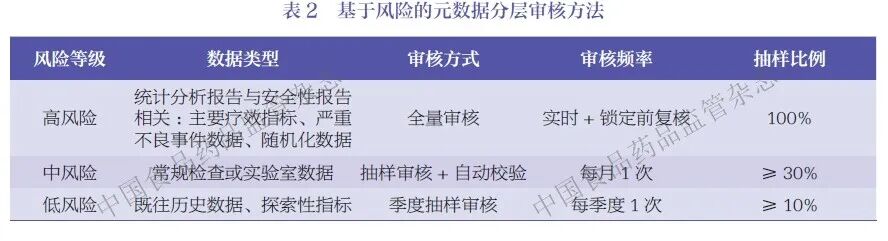
高风险环节需开展专项审核:①数据迁移/ 交换/ 映射环节(如随机化系统的随机入组信息同步到EDC 系统入组信息表)。审核映射关系文档与映射前后数据一致性校验报告、数据传输完整性效验(如 MD5 值比对结果)及传输加密稽查轨迹记录。②系统升级/ 变更环节。审核完整的变更控制记录(包括变更申请、风险评估、验证测试、确认报告、变更执行与变更关闭等全流程文件)、元数据标准调整方案及升级前后元数据一致性验证报告。③数据库锁定/ 解锁环节。审核解锁审批流程、解锁期间操作轨迹、锁定前元数据完整性校验记录。例如,某项目在 EDC 系统的eCRF 版本升级后,通过元数据审核发现3 条逻辑核查规则未同步升级迁移,经及时修复避免了数据核查遗漏;某疫苗临床试验中,EDC 系统迁移前验证不充分,系统升级导致旧版数据中元数据(如初始录入时间)丢失。上述两例问题的核心原因均为系统变更与数据迁移过程验证不充分,未开展系统性的元数据影响评估,迁移与升级范围未覆盖元数据一致性校验,最终导致元数据缺失或逻辑规则不同步。
2.3 特殊场景审核:盲态数据审核专项要求
盲态保护是临床试验数据完整性的核心要求,盲态相关元数据需采取专项保护措施,审核需聚焦“权限隔离、信息隐藏、操作追溯”3 大核心,确保临床试验的随机性与盲态不受破坏,避免因元数据泄露导致试验结果偏倚。
2.3.1 盲态相关元数据范畴
盲态保护覆盖的元数据需精准界定,包括 4 类核心信息:① 随机化敏感元数据,包括随机序列生成参数、区组大小等直接决定受试者分组逻辑的关键信息,属于核心盲态保密内容;分层因素(如年龄、性别、疾病分层等)仅为随机化设计信息,不属于敏感盲态元数据,无需纳入高强度保密范围。②治疗分组映射元数据,如受试者给药代码、药物编号与组别映射关系、药品标签编码规则等,为盲态保护的核心敏感信息。③非盲操作轨迹元数据,包括非盲人员账号、非盲模块访问记录、操作时间戳等,用于完整追溯非盲权限使用轨迹。④揭盲相关元数据,包括紧急揭盲申请单编号、揭盲原因、审批流程、执行时间及揭盲后数据处理记录等,用于规范紧急揭盲全流程并实现可追溯。同时,随机和试验供应管理系统(randomization and trial supply management,RTSM)版本、系统配置及验证记录属于系统级元数据,虽不直接泄露分组信息,但其完整性与合规性直接影响盲态可靠性,应纳入元数据审核范围。
2.3.2 盲态元数据审核实操路径
(1)权限隔离审核。重点审核以下几点:①核查非盲人员权限清单与试验方案、盲态管理规程的一致性,非盲角色需严格限定(如紧急揭盲授权人员、指定药物管理人员、独立非盲医学人员),相关权限申请、审批表编号及审批记录均应完整留存于元数据中。②验证权限分配遵循最小必要原则,如非盲临床药理人员仅可访问治疗分组相关信息,不得拥有临床数据修改权限。③通过定期访问日志与权限变更轨迹元数据核查权限使用合规性,重点监控盲态人员是否违规访问非盲模块(如盲态研究者账号多次触发非盲字段访问请求),或盲人员存在超范围访问行为。例如,某双盲Ⅲ期临床试验中,通过权限元数据审核发现1 名盲态人员账号被误授非盲模块访问权限,该账号曾查看3 例参与者治疗分组信息,项目组及时采取数据隔离、账号权限重新配置以及必要的补救措施,有效避免了试验偏倚进一步扩大。
(2) 信息隐藏审核。重点审核以下内容:①核查随机化元数据的加密存储状态,如随机分配序列需采用高强度加密算法[ 如高级加密标准(advanced encryption standard,AES)-256] 存储, 加密密钥由专人保管,元数据中需记录密钥生成时间、保管人信息及使用登记记录。②审核盲态元数据的展示形式,如EDC 系统中仅显示“试验药物A”“ 试验药物B” 等盲态标识,真实药物名称与组别映射关系在元数据中予以隐藏,避免通过字段注释、数据属性、质疑报告以及稽查轨迹等造成间接泄露。③验证盲态数据的传输安全性,盲态元数据传输应采用加密通道[ 虚拟专用网络(virtual private network,VPN)+ 安全套接层(secure sockets layer,SSL)双重加密],传输日志中需记录传输时间、接收方信息及完整性校验结果,防止数据在传输过程中被窃取或中断。例如,某肿瘤药物临床试验中,经元数据审核发现随机化参数在系统配置文档中以明文形式存储,项目组对其实施加密处理并补充加密相关日志后,以满足盲态保护相关合规要求。
(3)操作追溯审核。重点审核以下内容:①揭盲相关元数据需形成完整可追溯闭环,紧急揭盲仅可在方案规定的医学必要场景下实施(如处理严重不良事件、挽救危及生命的临床状况),元数据中需完整记录“申请- 评估-审批- 执行”全流程,无规范审批记录的揭盲行为均视为违规操作。②盲态相关元数据的修改应执行双人审批与授权机制,如随机化参数调整需经统计负责人与医学负责人双重审批确认,审批意见、操作人及时间戳等信息需完整留存于元数据中。③揭盲时序需与项目进度严格匹配,临床试验整体揭盲应在数据库锁定及统计分析方案定稿后执行;元数据需完整记录揭盲申请人、申请时间、审批人、审批时间、揭盲执行时间、操作人以及揭盲后数据访问轨迹,严禁提前揭盲或无依据揭盲。例如,某心血管药物临床试验中,通过揭盲元数据审核发现1 例非医学紧急场景下的违规揭盲(参与者因出现轻微头晕即启动揭盲),该操作无相应审批记录,且揭盲后未对相关数据采取隔离与控制措施,最终被判定为重要方案违背,纳入了监管报备。
2.3.3 盲态元数据常见风险与防控措施
本文对盲态元数据审核中常见的风险以及防控措施进行了总结,见表3。

3 元数据审核的实施流程与操作工具
3.1 审核实施流程
元数据审核需遵循“计划制定- 审核执行- 问题处理- 资料归档”全流程闭环管理原则,确保审核工作标准化、可追溯、可核查,具体实施步骤如下。
3.1.1 计划制定阶段
①申办者需在DMP 中明确元数据审核范围、风险分层标准、审核频率与责任分工,其中临床数据管理员负责试验级元数据审核,技术支持部门负责系统级元数据审核, 质量控制(quality control,QC)人员负责审核工作的监督与核查。②制定《元数据审核标准操作规程》,细化审核流程、验证方法、缺陷判定标准(如关键元数据缺失视为严重缺陷)及问题处理时限要求。③设计风险评估矩阵与标准化审核清单,明确数据全生命周期阶段元数据的审核要点(如数据采集阶段需审核设备校准记录、EDC 系统验证记录及数据录入人员权限资质等),并明确各审核要点所需的佐证材料(如设备校准证书、权限审批表、系统验证报告等)。④明确审核人员及资质要求(如具备临床试验数据管理相关经验、熟悉计算机化系统操作、掌握元数据审核相关规范等),并对审核人员开展专项培训,内容包括但不限于盲态保护要求、审核工具使用方法、缺陷判定标准等。例如,某申办者在Ⅲ期临床试验启动前,制定元数据专项审核计划,确定高风险元数据12 项、中风险23 项,并明确各风险等级的审核频率与验证方法,为后续审核工作的有序开展奠定了坚实基础。
3.1.2 审核执行阶段
①系统自动校验。依托EDC系统、LIMS 系统及元数据管理平台的内置校验功能,自动核查元数据时间戳一致性、逻辑核查规则匹配性,对缺失元数据、异常元数据进行自动预警(如设备校准周期过期、权限未及时注销等场景自动提示),同步生成系统自动校验报告,明确预警信息、校验结果及异常明细。②人工抽样复核。采用风险分层审核原则,对高风险元数据实施100% 全量审核、中风险元数据抽样比例不低于 30%、低风险元数据抽样比例不低于 10% ;审核人员需逐一核对元数据及佐证材料,规范填写《元数据审核记录表》,完整记录审核结果、发现的缺陷问题、佐证材料编号及审核时间戳。③专项审核。针对数据迁移、系统升级、数据库锁定/ 解锁、揭盲等关键高风险环节,组建跨部门联合审核小组(由临床数据管理、技术支持、质量控制等相关部门人员组成),按照专项审核流程开展全面审核,重点核查环节合规性、元数据一致性及风险防控落实情况,审核完成后形成专项审核报告,明确审核结论、问题及整改建议。例如,某多中心临床试验在数据迁移完成后,通过自动校验发现8 条数据映射关系异常,审核团队随即开展人工复核与跨部门联合审核。最终确认异常原因系系统接口参数设置错误,经及时修复并重新校验后,确保了元数据及原始数据的完整性与准确性。
3.1.3 问题处理阶段
①建立元数据问题追踪系统。对审核发现的问题按严重程度实施分级管理(轻微缺陷、一般缺陷、严重缺陷),系统需完整记录问题编号、发现时间、问题详细描述、责任部门、责任人、整改措施、整改时限及整改验证结果,确保问题可追溯、可核查。②明确分级整改要求。轻微缺陷(如元数据格式不规范、备注信息不完整)需在3 个工作日内完成整改;一般缺陷(如非关键元数据缺失、填写不规范)需在7 个工作日内补充完善;严重缺陷(如稽查轨迹缺失、盲态信息泄露、关键元数据篡改)需立即启动风险评估,暂停相关临床试验流程,同步上报质量部门及项目负责人,必要时上报监管机构。③整改验证与闭环管理。审核团队需对整改结果进行专项复核,确保问题整改到位、形成闭环,如设备校准元数据缺失的,需补充完整校准证书,并验证校准结果的有效性及校准记录的规范性。例如,某临床试验审核发现10 例参与者数据更正未记录修改原因(判定为一般缺陷),责任部门在5 个工作日内补充了修改说明及原始记录相关元数据,经审核团队复核确认,整改结果符合元数据审核规范及试验方案要求。
3.1.4 资料归档阶段
①相关记录需以不可编辑格式(如PDF/A 标准格式)归档,归档内容需包含审核人签字、审核日期、审核结论、问题清单、整改报告、验证记录等完整信息,确保归档记录的真实性与完整性。②归档元数据需与对应的原始数据、试验文档建立唯一关联,保障全流程可追溯(如稽查轨迹元数据与参与者eCRF 数据通过唯一参与者标识符关联,确保数据溯源可查)。③归档期限需严格符合监管要求,至少保留至药品上市后5 年或临床试验终止后10年(以期限长者为准)。归档存储需采用“本地+ 异地”双备份模式,定期开展备份恢复测试,确保归档数据的安全性、可读性及可访问性。例如,某申办者通过搭建元数据归档管理系统,实现审核记录、元数据及佐证材料的电子化归档、分类管理与快速检索,在监管核查过程中大幅提升了核查效率,确保核查过程顺畅、可追溯。
3.2 常用操作工具与技术支撑
元数据审核的高效实施需依托适配的工具与技术支撑,结合临床试验元数据审核场景及监管要求,常用工具可分为基础工具与进阶工具两类,分别满足常规审核与高复杂度审核需求。
3.2.1 基础工具
①风险评估工具。如Excel风险矩阵模板、在线风险评估平台等,支持按数据重要性、系统复杂度、操作风险等维度量化风险等级,明确风险权重,自动生成标准化风险清单及风险评估报告,为审核重点分配提供依据。②标准化审核清单。按数据全生命周期阶段(采集、处理、传输、存储、运维、销毁)与元数据类型(技术元数据、过程元数据、业务元数据)分类设计,明确包含审核要点、缺陷判定标准、佐证材料要求及备注栏,确保审核工作标准化、无遗漏。③问题追踪工具。如系统缺陷数据库(Bug Database)、项目管理工具等,支持元数据审核问题的创建、分配、分析、跟踪、整改、验证、关闭全流程闭环管理,可设置整改时限自动提醒功能,同步自动生成问题统计报表(含缺陷分级、整改完成率、责任部门分布等)。④文档管理工具。使用电子文档管理系统,实现元数据审核计划、标准操作规程、审核记录、整改报告、验证报告等相关文档的集中存储、版本控制、权限管理及检索查询,确保文档可追溯、可核查。
3.2.2 进阶工具
① 专业级的元数据管理平台。如Informatica Metadata Manager、IBM InfoSphere 等主流平台,支持元数据自动采集、分类、关联及可视化分析,可实现跨系统元数据统一管控。例如,自动识别EDC 系统与LIMS 系统的元数据映射关系,精准排查映射偏差,提升跨系统元数据管理效率[21-24]。② 稽查轨迹分析工具。如Oracle Audit Vault、EDC 系统内置稽查轨迹导出与可视化模块,支持按操作人、时间范围、数据类型、操作类型等维度筛选分析,可自动识别异常修改行为。例如,集中批量修改数据、频繁修改关键元数据,为违规操作核查提供技术支撑[25]。③一致性校验工具。如MD5 校验工具、WinSCP 文件传输校验功能等,可精准验证数据迁移、传输过程中的元数据与原始数据完整性,自动生成校验报告,明确校验结果、异常明细及完整性判定依据。④智能化审核工具。基于人工智能(artificial intelligence,AI)算法构建异常识别模型,实现异常元数据自动预警,如精准识别时间戳逻辑异常(如操作时间早于数据创建时间、时间戳重复)、权限变更异常(短期内频繁调整同一用户权限、违规授予高风险权限) 等潜在问题。例如, 笔者团队搭建的临床数据全流程管理系统(clinical datatotal management system,CDTMS),实现了元数据审核的标准化、自动化与规范化管理,经近百个项目的实践验证,该系统可将审核效率提升40%,异常问题发现率提升30%,大幅降低了人工审核成本与人为误差[26]。
3.2.3 工具选型原则
①合规性原则。工具须具备数据加密、操作全程追溯、电子记录不可篡改等核心功能,完全满足相关文件对临床试验电子记录的相关要求,确保审核过程与结果可核查、可追溯。②适配性原则。结合临床试验规模、数据量及系统复杂度合理选型,中小型临床试验可采用基础工具组合(如 Excel 风险矩阵+ 问题追踪工具),满足常规审核需求;大型多中心临床试验、复杂系统集成场景,建议选用专业化元数据管理平台,提升审核效率与管控精度。③易用性原则。工具操作界面应简洁直观,支持审核记录导出、标准化报表自动生成、数据检索等实用功能,降低审核人员学习成本,确保工具可快速落地应用。④扩展性原则。工具需支持与现有临床试验计算机化系统(如EDC、LIMS、RTSM 等)无缝对接,实现元数据自动同步与共享,同时可适应临床试验流程变更、系统升级等需求,保障工具长期适配性。
4 元数据审核的关键注意事项
4.1 盲态保护的额外注意事项
①盲态试验中,盲态相关元数据应存储于独立加密服务器,并与非盲数据实现物理或逻辑隔离;访问权限实行“双人双钥”管理,每次访问均需记录操作目的[ 如统计分析使用、 严重不良事件(serious adverse event,SAE) 核查使用],形成完整访问轨迹。②系统配置阶段应屏蔽非盲人员操作界面中的盲态元数据字段,如隐藏药物编号、分组映射等敏感属性描述、禁用非盲模块入口,防止通过查看页面源代码、导出数据字典等方式间接泄露盲态信息。③揭盲后应开展盲态保护专项元数据审核,全面核查盲态元数据的访问记录、修改痕迹与操作日志,确认无违规访问与不当操作,并形成盲态保护完整性总结报告。例如,某双盲临床试验在揭盲后经元数据审核发现,1 名盲态人员在试验期间违规访问3 例参与者分组元数据,经内部质量核查判定为严重违规,项目组及时采取整改与防控措施,未对试验整体造成重大影响。
4.2 数据隐私保护与合规要求
①元数据中包含的个人敏感信息(如参与者身份证号、姓名、住址、出生日期等),应按照隐私保护相关法律法规要求实施脱敏处理。②跨境传输的元数据需符合数据出境安全评估相关规定,不得携带敏感个人信息及国家核心数据;传输前应完成数据安全评估,并在元数据体系中留存评估报告编号与授权传输记录。③元数据的存储与访问严格遵循“最小必要原则”,仅授权人员可访问与其岗位职责相关的元数据内容,避免信息过度采集与非必要暴露[27-29]。例如,某跨境多中心临床试验因未对元数据中的参与者出生日期等信息脱敏,导致数据出境审核流程受阻、项目进度延误。
4.3 第三方管理的注意事项
① 申办者应在与CRO、SMO、中心实验室等第三方机构签订的合同及协议中,明确约定元数据审核相关要求与责任边界,包括审核范围、审核频率、报告提交格式与时限,以及第三方配合申办者开展监查、稽查与核查的义务。②对第三方系统元数据进行审核时,应重点验证系统的合规性(如是否完成计算机化系统验证)、数据传输协议的标准化程度[ 如是否采用Health Level Seven Fast Healthcare Interoperability Resources Release 4(HL7 FHIR R4)标准] 以及元数据完整性( 如实验室设备校准记录、数据传输加密日志、访问审计轨迹等)。③建立第三方元数据质量考核与问责机制,将元数据审核结果纳入第三方绩效评价体系,对存在严重数据缺陷或持续不合规的第三方,采取限制整改、暂停业务合作乃至终止合作等处置措施。④第三方人员调岗或离职时,应同步审核其账号注销记录与权限回收等相关元数据,确保账号及时失效或禁用,防范账号复用、越权访问等数据安全风险。例如,申办者在审核某研究中心LIMS系统元数据时,发现设备校准记录已过期且未及时更新与补正,申办者立即对该实验室采取暂停数据接收、限期整改等措施,必要时启动实验室更换流程,有效保障了临床试验源数据与元数据质量。
4.4 跨部门协作与人员能力保障
①建立健全跨部门协作机制。明确临床数据管理、IT/ 技术支持、统计、质量、医学、监查等相关部门的职责分工:临床数据管理部门负责试验级元数据审核、IT/ 技术支持部门负责系统级元数据管控、质量部门负责审核过程的监督与核查、统计部门参与衍生数据相关元数据的审核与验证,各部门协同配合,形成“分工明确、责任到人、协同高效” 的管理闭环。②开展常态化、分层分类培训。培训内容涵盖元数据定义与范畴、各生命周期审核要点、盲态保护规范、审核工具操作方法、相关法规文件解读、异常问题识别与处理等。结合岗位履职需求设计专项培训,如针对研究者,培训重点聚焦元数据浏览规范与合规要求;针对IT 人员,培训重点聚焦系统级元数据采集、管理与异常排查。③建立审核人员资质认定与持续能力保障机制。审核人员需通过理论考试与实操考核,取得相应资质后方可上岗;定期开展能力评估与再培训,及时更新审核知识与技能,确保审核人员能力持续符合元数据审核工作要求。此外,还应建立跨部门协作小组,完善定期培训与沟通机制,强化各部门协同配合,有效降低元数据审核缺陷率,提升审核质量与效率。
4.5 系统变更与适配的注意事项
(1)计算机化系统升级、补丁修复或试验方案修订时,需同步更新元数据审核范围与审核标准,提前开展变更影响分析及风险评估,元数据中需完整留存变更控制记录、影响/ 风险评估报告及验证报告。此外,还应明确变更对元数据类型、审核要点、风险等级的具体影响,如试验方案新增生物标志物指标时,需同步补充该指标对应的业务元数据(数据点定义、逻辑核查规则、编码标准等)及过程元数据(稽查轨迹、访问权限配置等)的审核要求,避免因变更遗漏导致元数据审核出现盲区。例如,某临床试验项目在试验方案变更时,新增了研究中心实验室检测项目,相关数据批量导入到EDC 系统,但未同步更新该批量导入功能对应的元数据审核标准(包括批量导入操作的时间戳、操作人、数据来源校验记录、完整性校验结果等),导致后续元数据审核中发现3 批导入数据缺少来源追溯相关元数据,需重新补充开发功能并完成验证,造成项目进度延误。
(2)系统升级后需开展元数据一致性验证,重点核查变更前后元数据的连续性、兼容性及完整性,具体包括3 个维度:①技术元数据方面。确认系统升级后设备校准记录、数据存储路径、加密算法、系统版本等关键信息无异常变更。②过程元数据方面。验证稽查轨迹、访问日志的记录格式、核心要素(如操作人、时间戳、操作类型、修改原因)未发生缺失或异常。③业务元数据方面。确保逻辑核查规则、衍生数据计算公式、数据编码标准等未因系统升级出现偏差, 并与DMP 要求一致。例如,某多中心临床试验在更换实验室数据传输接口后,通过元数据一致性验证发现,数据映射关系缺失2 个关键字段(含样本采集时间与检测时间),项目组及时补充映射规则并重新验证,避免了数据传输错误及元数据缺失风险。
(3)试验过程中若涉及系统更换(如从一个EDC 系统迁移至另一个EDC 系统),需开展全量元数据迁移验证,确保迁移过程合规、元数据完整可追溯,具体步骤如下:①提前制定专项元数据迁移方案,明确迁移范围、数据映射规则、验证方法、责任分工及时限。②迁移完成后,采用分层抽样对比方式开展验证(高风险元数据实施100% 全量比对,中、低风险元数据抽样比例不低于50%),以确认迁移前后元数据的完整性、准确性与一致性。③完整留存迁移过程相关元数据(迁移时间、操作人、校验结果、异常处理记录等),形成元数据迁移验证报告并归档,作为监管核查佐证材料。例如,某肿瘤药物临床试验中期,因系统功能需要变更EDC 系统,由于未开展元数据全量迁移验证,导致参与者的SAE 相关稽查轨迹部分丢失。项目组不得不重新从旧系统导出相关元数据归档,并补充缺失原因说明及验证记录,增加了额外工作量与监管风险。结合实际工作经验及监管要求,笔者不建议在临床试验已产生数据且处于运行阶段时,将项目从一个EDC系统迁移到另一个EDC 系统,若确需迁移,需提前开展充分的风险评估并严格执行全量验证流程。
(4)建立系统变更后的审核试运行机制,试运行期限建议不少于1 个月。试运行期间需提升元数据审核频率(如每周开展1次专项审核),重点监控变更相关元数据的异常情况,如系统升级后是否出现时间戳格式不一致、权限配置错乱、逻辑核查规则失效、元数据同步延迟等问题。试运行结束后形成试运行评估报告,根据试运行情况优化审核标准与流程,确保变更后的元数据审核工作符合相关监管要求及试验实际需求,避免因系统变更导致元数据缺陷。
5 临床试验元数据审核存在的问题
5.1 元数据定义与范畴不统一,审核范围模糊
当前,临床试验行业内尚未形成统一的元数据标准化体系,申办者、CRO 以及临床研究机构之间对元数据的定义、分类及核心清单存在差异。实践中,部分单位仅将稽查轨迹、设备校准记录等基础元数据纳入审核范围,遗漏了逻辑核查规则变更记录、数据传输映射关系、系统配置日志等关键元数据。同时,存在混淆试验级与系统级元数据审核责任的情况,导致系统级元数据(如服务器配置日志、数据库运维记录)长期未开展或未全面开展有效审核,形成审核盲区。此外,行业缺乏明确的元数据分类标准与审核清单,尤其在多中心临床试验中,因各研究中心对元数据定义与审核标准理解不一致,容易出现跨中心审核尺度不统一、审核结果缺乏可比性等问题,这也是当前元数据审核工作中值得重点关注的行业痛点。
5.2 盲态保护措施不完善,风险防控存在短板
盲态元数据的保护与审核是行业普遍薄弱环节,核心问题集中在权限配置、信息隐藏、流程管控3 个层面:①权限配置层面。部分临床试验未建立严格的“角色- 权限”映射机制与权限分级管控体系,存在盲态人员误获非盲权限的安全风险。例如,某双盲临床试验中,曾出现医学经理通过系统权限漏洞违规查看随机化元数据的案例,严重违背盲态保护原则。②信息隐藏层面。部分盲态敏感元数据(如药物编号与组别映射关系、随机序列参数)未采取高强度加密存储,仅通过系统界面隐藏,存在通过后台配置文件获取盲态信息的风险,无法形成有效的盲态防护屏障。③流程管控层面。部分临床试验未建立揭盲相关元数据全流程闭环管理机制,存在无审批记录直接揭盲、揭盲后未及时对相关数据采取隔离控制、未完整记录揭盲操作轨迹等违规行为,直接影响临床试验结果的客观性与可靠性。
5.3 第三方管理薄弱,责任边界不清
在多中心临床试验中,第三方机构(包括CRO、SMO、中心实验室等)参与度高,但其元数据审核管理薄弱、责任边界界定模糊等问题突出,具体表现为3 个方面:①合同约定层面。大部分申办者在与第三方签订的合作合同中,未明确元数据审核的具体要求、频率、报告格式及责任划分,导致部分CRO 未按规范提交元数据审核报告,或报告内容流于形式、缺乏实质审核内容,无法真实反映元数据质量。②审核管控层面。申办者对第三方系统元数据的审核力度不足,未有效验证第三方系统的合规性(如LIMS 系统设备校准记录真实性、数据传输加密合规性)。例如,某临床试验中,曾发现中心实验室缺乏有效的设备校准元数据,导致相关检测数据可靠性存疑,影响试验数据完整性。③人员管理层面。第三方人员离职后账号未及时注销、系统访问权限未彻底回收的情况时有发生,导致第三方离职人员仍可违规访问临床试验元数据的安全隐患,给数据隐私与盲态保护带来风险。
5.4 技术工具适配不足,审核效率与准确性偏低
临床试验元数据具有数量大,类型杂、审核内容繁琐等特点,当前元数据审核仍以人工操作为主,技术工具支撑不足的问题较为突出,主要体现在3 个方面:①基础工具依赖严重。中小型申办者多依赖Excel、Word 等基础办公工具开展元数据审核,此类工具缺乏审核报告的自动化生成、数据完整性校验、异常元数据自动预警等功能,不仅导致审核效率低下,还易因人为疏忽遗漏元数据异常,如时间戳异常、权限配置错误等。②工具兼容性较差。部分机构使用的元数据审核工具与现有计算机化系统(如EDC、LIMS、RTSM 等)兼容性不足,无法实现元数据自动采集、同步与比对,需人工录入核对元数据,大幅增加了数据录入错误风险,也降低了审核工作的连贯性。③智能化工具应用滞后。行业内鲜少有申办者采用基于AI的异常元数据识别工具,难以快速识别批量修改数据、时间戳逻辑矛盾、权限频繁变更等隐蔽性强、人工难以察觉的问题,影响了元数据审核的质量与效率。
5.5 人员能力不足,审核专业性欠缺
元数据审核对人员的综合能力要求较高,需同时具备临床数据管理、计算机化系统操作、相关法规理解等专业能力,但当前行业内相关人才储备不足,人员专业能力参差不齐,主要表现为3 个方面:①专项培训缺失。大部分元数据审核人员未接受系统的元数据审核专项培训,对元数据的分类标准、审核要点、盲态保护要求、合规法规等理解不深入、不全面,审核工作存在“重形式、轻实质”的现象,仅核对表面信息,未开展实质性的风险核查。②专业知识薄弱。部分审核人员缺乏计算机化系统相关知识,无法识别系统级元数据(如数据库备份日志、接口通信协议、加密算法配置)的异常情况,难以发现系统层面的元数据缺陷与安全风险。③人员流动性大。临床试验项目周期长,而元数据审核人员流动性较高,部分项目因审核人员离职,导致审核工作中断、审核标准执行不一致,严重影响了元数据审核的连续性、规范性与完整性。
6 优化对策与思考
6.1 建立标准化元数据体系,明确审核范围
(1)依托国内外监管核心要求, 建议相关部门可参考《E6(R3):药物临床试验质量管理规范技术指导原则》《药物临床试验计算机化系统和电子数据指导原则(征求意见稿)》等文件要求,并联合行业协会制定“临床试验元数据标准化相关指南”,统一元数据的定义、分类、核心清单及审核标准,明确试验级与系统级元数据的审核责任分工。建议将系统验证记录、稽查轨迹、设备校准记录、权限变更日志、随机化与盲态元数据、逻辑核查规则、数据传输映射关系等纳入强制审核清单,确保无核心遗漏,满足日益提高的监管要求[30-32]。
(2)申办者需在DMP 中细化元数据审核相关内容,结合试验特点制定个性化的元数据分类标准与风险清单,明确各类型元数据的审核频率、抽样比例、判定标准。跨中心临床试验需统一各中心的审核标准与流程,避免因标准差异导致的数据质量不均。例如,某国际多中心临床试验的申办者通过采用标准化元数据体系,跨中心元数据审核差异率明显降低,显著提升了数据一致性[33]。
6.2 强化盲态保护技术与流程,筑牢风险防线
(1)技术层面。主要包括:①盲态元数据采用“加密存储+物理隔离” 模式, 使用AES-256 等高强度加密算法,加密密钥实行“双人双钥”管理,定期更换密钥。②系统配置时屏蔽盲态用户界面的盲态元数据字段,禁用后台配置文件访问权限,从技术上杜绝信息泄露隐患。③跨系统传输盲态元数据时, 采用VPN+SSL 双重加密通道,传输后自动校验数据完整性,确保传输过程安全可控。
(2)流程层面。重点涵盖:① 建立严格的非盲权限审批机制,非盲角色需在方案中明确界定,权限申请需经申办者的项目负责人审批,相关审批记录完整留存于元数据体系中。②系统强制关联揭盲审批表,无有效审批编号则无法执行揭盲操作,揭盲后自动触发数据隔离控制与操作日志记录,实现揭盲流程全追溯。③定期开展盲态元数据专项审计,重点核查权限使用日志、盲态元数据访问记录,及时发现并处置违规行为,防范盲态泄露风险。
(3)培训层面。针对盲态保护要求开展专项培训,覆盖所有参与临床试验的相关人员(研究者、监查员、数据管理员、统计师等),强化全员盲态保护意识,明确违规操作的后果与责任,降低人为失误引发的盲态保护风险。
6.3 规范第三方管理,明确责任边界
(1)合同约束。申办者在与第三方签订的合作合同中需明确元数据审核要求,包括审核范围、频率、报告格式与时限,以及第三方配合申办者开展监查、稽查的义务。将元数据审核结果纳入第三方绩效评估体系,对存在严重缺陷的第三方采取整改、暂停合作、终止合同等分级处置措施,强化第三方责任意识。
(2)审核管控。主要包括:①建立第三方系统元数据合规性验证机制,合作前验证第三方系统的元数据采集、存储、传输功能是否符合监管要求,合作期间按季度开展专项审核,持续监控第三方元数据质量。②要求第三方提交完整的元数据审核报告,包括审核记录、问题清单、整改措施及验证结果,申办者按比例抽样复核(高风险项实施100%复核),确保审核报告的真实性与完整性。③建立第三方人员权限动态管理机制,要求第三方每月提交人员变动与权限调整清单,申办者同步审核账号注销与权限回收相关元数据,防范人员变动引发的数据安全风险。
(3)协同机制。搭建申办者与第三方的元数据共享平台,实现元数据实时同步与问题协同处理。定期召开第三方元数据管理沟通会,通报审核情况,协调解决合作中的难点问题,形成“合同约束- 审核管控- 协同推进”的管理闭环,提升审核效率与质量[34]。
6.4 推广先进技术工具,提升审核效能
(1)分层适配工具。中小型申办者可采用“基础工具组合”(风险矩阵模板+ 审核列表+ 问题追踪系统),在控制工具投入成本的同时,满足常规元数据审核需求。大型多中心临床试验、复杂系统集成场景,建议选用专业化元数据管理平台,实现元数据自动采集、分类、关联与可视化分析,大幅提升审核效率与管控精度。
(2) 强化工具适配性。选择与现有计算机化系统(EDC、LIMS、RTSM 等)兼容的审核工具,实现元数据自动同步与接口无缝对接,减少手动录入工作量,降低数据录入错误风险。审核工具需具备数据加密、操作全程追溯、电子记录不可篡改等合规功能,满足相关法规文件对电子记录的要求。
(3)推进智能化应用。推广基于AI 的异常元数据识别工具,通过算法模型自动识别时间戳异常、批量修改数据、权限变更异常等隐蔽问题,提升异常问题发现率。探索区块链技术在元数据存储中的应用,实现元数据全生命周期不可篡改追溯,进一步保障数据可靠性与可核查性[35]。
6.5 加强人员能力建设,提升审核专业性
(1)建立系统化培训体系。培训内容应涵盖元数据定义与分类、审核要点、盲态保护要求、审核工具操作、相关法规文件解读等,针对不同岗位(临床数据管理员、技术支持人员,IT、监查员)设计专项培训模块,贴合岗位履职需求。采用“理论+ 实操”相结合的培训方式,实操案例应涵盖盲态元数据审核、系统变更后元数据验证、第三方元数据审核等场景,确保培训效果落地。
(2)建立资质认定与持续评估机制。审核人员需通过理论考试与实操考核,取得相应资质后方可上岗;定期(建议每年度)开展能力评估,评估不合格者暂停审核工作,参加再培训后重新考核,合格后方可恢复上岗。鼓励审核人员考取相关专业资质,提升专业素养。
(3)搭建行业交流平台。行业协会可组织元数据审核经验交流会、案例研讨会,分享优秀实践与典型违规案例,促进行业整体审核能力提升。鼓励申办者、CRO、监管机构开展合作研究,共同解决元数据审核中的技术难点与合规问题,推动行业元数据审核水平持续进步。
6.6 规范元数据销毁管理,闭环全生命周期合规
作为元数据全生命周期的最终环节,销毁管理需围绕“ 审批合规、过程可溯、效果可验” 构建标准化流程,填补全生命周期管理空白,避免因销毁不当引发数据泄露或监管风险。
(1)建立销毁前审批机制,明确元数据销毁的“ 准入门槛”:①制定元数据销毁管理标准操作规程,明确销毁申请需提交待销毁元数据清单(含数据类型、存储位置、关联试验信息)、销毁理由(如超留存期限、数据冗余、试验终止)、拟销毁时间、拟销毁方式等,清单需经数据管理负责人与质量部门双重审核。②强化留存期限核查,依据相关要求,确认待销毁元数据已满足药品上市后 5 年或试验终止后 10 年(取较长者)的最低留存期限;高风险元数据(如随机化方案、严重不良事件关联元数据)需额外经医学负责人审批,杜绝未达期限违规销毁。③留存审批全流程元数据,包括审批人资质证明、审批意见、审批时间戳,确保审批链路可追溯[36]。
(2)规范销毁过程操作,确保每一步留痕可查:①明确销毁操作主体资质,仅授权“ 数据安全管理员+ 质量监督员” 共同执行销毁,操作前需通过系统身份验证,元数据中详细记录操作人账号、操作 IP 地址、终端设备唯一标识。②区分销毁方式制定操作标准:物理销毁(如硬盘、U 盘等存储载体)需委托具备资质的第三方机构,提供粉碎过程视频记录、第三方见证报告及残渣检测证明;电子销毁(如服务器数据、云存储元数据)需采用多次覆写(至少 3 次)+ 加密擦除技术,留存擦除算法日志(如AES-256擦除记录),云存储元数据需获取服务商出具的《销毁完成回执》。③实时记录销毁过程元数据,包括销毁开始/ 结束时间、销毁进度、异常情况处理记录(如销毁中断后的重启验证),形成《元数据销毁过程报告》[36]。
(3)强化销毁后效果验证,杜绝数据残留风险:①委托独立的第三方开展销毁效果验证,电子元数据需通过专业工具检测“ 是否存在可恢复数据片段”,物理载体需核查“ 是否彻底粉碎至无法复原”,验证结果需形成《销毁效果验证报告》。②核对“ 销毁前后数据清单”,确认待销毁元数据已全部移除,无遗漏或误销毁(如误删仍需留存的核心业务元数据),差异项需立即排查原因并整改。③归档全套销毁元数据,将“ 申请单、审批记录、过程报告、验证报告” 按监管要求归档至异地备份系统,留存至元数据销毁后5 年,确保监管核查时可完整追溯销毁全流程[36]。
7 结论
元数据作为临床试验数据全生命周期治理的“ 核心脉络”,是连接原始数据与监管合规的关键纽带,其审核工作的系统性与完整性直接决定临床试验数据质量与结果可信度。本文通过对元数据审核体系的全面分析,形成了以下核心成果。
在基础认知层面,明确了元数据与原始数据的本质差异,界定了试验级与系统级元数据的管理边界,细化了技术、过程、业务3类元数据的具体范畴,为行业统一元数据认知提供了参考。
在审核实操层面,确立了“ 范围全覆盖、内容全维度、对象分风险、实施全流程” 的核心原则,构建了包含全生命周期审核场景、“五性” 验证标准、风险分层机制的实操体系,尤其针对盲态数据这一高风险场景,提出了权限隔离、信息隐藏、操作追溯的三重保护方案,填补了行业特殊场景审核的实操空白。
在实施保障层面,规范了“ 计划- 执行- 问题处理- 归档” 的闭环流程,明确了基础与进阶两类工具的适配场景,梳理了盲态保护、隐私合规、第三方管理等关键注意事项,为审核工作落地提供了全方位支撑。针对行业现存的5 大突出问题,本文提出的标准化体系建设、技术工具升级与人员能力提升等思考,具有较强的实践指导意义。
未来,随着临床试验数字化、智能化的发展,元数据审核将向自动化、精准化、全流程追溯方向演进。行业需持续推动元数据标准化建设,强化技术与流程的深度融合,提升从业人员专业素养,不断完善审核体系。本文构建的实操框架可为行业高质量发展提供坚实支撑,助力临床试验数据质量提升,最终保障公众用药安全。
作者简介
颜崇超,博士,江苏恒瑞医药生物计量部临床数据科学中心,副总经理。专业方向:临床试验数据治理、临床数据管理、临床试验随机化与药品管理、临床试验一体化平台建设与计算机化系统验证
参考文献:略




编辑:李丹
审核:赵燕宜






