 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库周末文摘 | 《医疗器械生物学评价 第18 部分:风险管理过程中医疗器械材料的化学表征》的技术框架及可浸提物和可沥滤物分析


引用本文
卢忠,陈德羡,刘贻声,张广湘,王春仁,孔繁圃*.《医疗器械生物学评价 第18 部分:风险管理过程中医疗器械材料的化学表征》的技术框架及可浸提物和可沥滤物分析[J].中国食品药品监管,2026(4):84-93.
《医疗器械生物学评价 第18 部分:风险管理过程中医疗器械材料的化学表征》的技术框架及可浸提物和可沥滤物分析
Technical Framework of Biological Evaluation of Medical Devices - Part 18: Chemical Characterization of Medical Device Materials within a Risk Management Process, and Analysis of Extractables and Leachables
卢忠
中国药品监督管理研究会医疗器械监管研究专业委员会
LU Zhong
Special Committee for Medical Device Regulation, China Society for Drug Regulation
陈德羡
北京智慧医疗技术创新联盟医疗器械创新与非临床研究专业委员会
CHEN De-xian
Special Committee on Medical Device Innovation and Non-Clinical Research, Beijing Wise Information Technology of Med Industry Innovation Alliance
刘贻声
北京智慧医疗技术创新联盟医疗器械创新与非临床研究专业委员会
LIU Yi-sheng
Special Committee on Medical Device Innovation and Non-Clinical Research, Beijing Wise Information Technology of Med Industry Innovation Alliance
张广湘
北京智慧医疗技术创新联盟医疗器械创新与非临床研究专业委员会
ZHANG Guang-xiang
Special Committee on Medical Device Innovation and Non-Clinical Research, Beijing Wise Information Technology of Med Industry Innovation Alliance
王春仁
北京智慧医疗技术创新联盟医疗器械创新与非临床研究专业委员会
WANG Chun-ren
Special Committee on Medical Device Innovation and Non-Clinical Research, Beijing Wise Information Technology of Med Industry Innovation Alliance
孔繁圃*
清华大学医学院
KONG Fan-pu*
Tsinghua Medicine, Tsinghua University
摘 要 / Abstract
GB/T 16886.18—2022《医疗器械生物学评价 第18 部分:风险管理过程中医疗器械材料的化学表征》是我国医疗器械生物学评价的核心标准之一。本文介绍了该标准的技术框架,包括信息收集、材料和医疗器械的生物学等同性判定和多层次化学表征,并特别讨论了可浸提物和可沥滤物分析的关键技术环节,介绍了分析评价阈值在未知可沥滤物筛选中的作用。科学、规范的化学表征能够为医疗器械的毒理学风险评估提供坚实的基础,提升生物学评价的效率,减少不必要的动物试验,有力保障医疗器械的生物相容性。
GB/T 16886.18—2022 Biological Evaluation of Medical Devices - Part 18: Chemical Characterization of Medical Device Materials within a Risk Management Process is a core standard for biological evaluation of medical devices in China. This paper outlines the technical framework of the standard, including information gathering, assessment of biological equivalence for materials and devices, and multi-level chemical characterization. Special emphasis is placed on the key technical aspects of extractables and leachables (E&L) analysis, including the application of analytical evaluation threshold (AET) in screening non-targeted leachables. Scientifically sound and standardized chemical characterization provides a robust foundation for toxicological risk assessment, enhances the efficiency of biological evaluation, reduces unnecessary animal testing, and ultimately ensures the biocompatibility of medical devices.
关 键 词 / Key words
医疗器械;生物学评价;化学表征;可浸提物;可沥滤物
medical devices; biological evaluation; chemical characterization; extractables; leachables

GB/T 16886《医疗器械生物学评价》系列标准是我国医疗器械生物学评价领域的基础性技术标准。该系列标准以GB/T16886.1—2022《医疗器械生物学评价 第1 部分:风险管理过程中的评价与试验》[1] 为总纲,规定了医疗器械生物学评价的核心原则和要求;围绕生物学评价,GB/T16886 细分出化学表征(GB/T16886.18—2022《医疗器械生物学评价 第18 部分:风险管理过程中医疗器械材料的化学表征》[2])、毒理学风险评估(GB/T16886.17—2025《医疗器械生物学评价 第17 部分:医疗器械成分的毒理学风险评估》[3])、样品制备与参照材料(GB/T16886.12—2023《医疗器械生物学评价 第12 部分:样品制备与参照材料》[4])、各类生物学试验方法等专项标准,覆盖医疗器械生物学评价的多个环节。
过去30 年间,医疗器械生物学评价体系经历了显著的范式转变。传统的医疗器械生物学评价主要依赖细胞或动物试验。这些试验虽然具有很高的参考价值,但未能识别医疗器械释放的化学物质,无法在分子等微观层面揭示材料和生物体的相互作用机制。随着分析化学技术的发展和毒理学数据的积累,医疗器械领域相关从业者逐渐认识到,通过化学表征和毒理学风险评估相结合的方式,能够利用最少必要信息,高效完成医疗器械的生物学评价。早期的GB/T 16886.1—2001《医疗器械生物学评价第1 部分:评价与试验》[5] 突出“ 动物试验主导”;而GB/T16886.1—2011《医疗器械生物学评价 第1 部分:风险管理过程中的评价与试验》[6] 和现行标准GB/T 16886.1—2022[1] 已将化学表征前置,对于刺激性、全身毒性、遗传毒性、生殖毒性、发育毒性、致癌性等毒理学终点,强调化学表征先行,在化学表征的基础上结合已有数据开展毒理学风险评估,必要时补充生物学试验和毒代动力学研究,最后完成生物学评价。
GB/T 16886.18—2022[2] 是我国现行的医疗器械材料化学表征的专项标准。本文立足“表征先行”的理念,系统讨论GB/T16886.18—2022 的技术框架,并重点介绍可浸提物和可沥滤物分析,以期为行业开展化学表征工作、落实风险管控要求提供技术参考。

01
GB/T 16886.18—2022 标准框架
GB/T 16886.18—2022[2] 中关于医疗器械材料化学表征的流程见图1。本标准构建了信息收集、生物学等同性判定、多层次化学表征的实施流程,每个环节层层递进。需要注意的是,该流程设有多个进入点和退出点,分析人员不需要完成所有步骤,而应当根据实际情况灵活选择,合理利用最少必要信息,科学地完成化学表征过程。
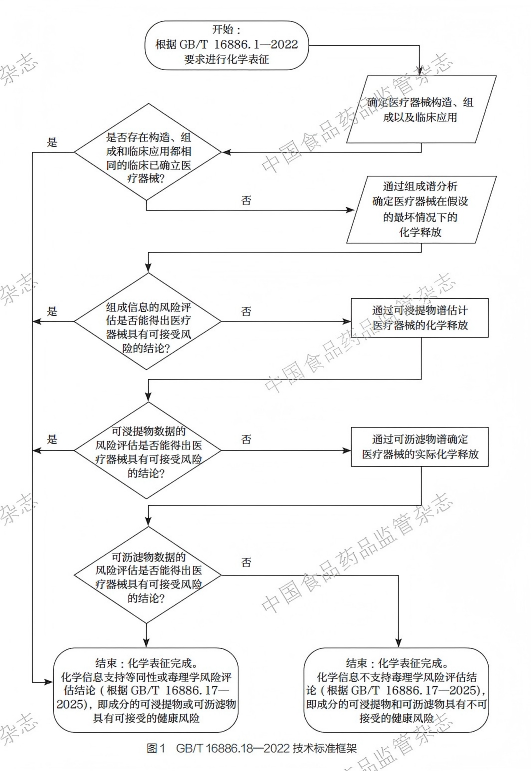
1.1 信息收集
化学表征的首要步骤是收集和整合医疗器械的信息。GB/T16886.18—2022[2] 强调信息收集需覆盖产品全链条,其内容包括:①医疗器械的用途、人体接触性质、接触时长、适用人群。②产品材料构造和组成、添加剂、工艺残留物、产品成分降解特性、灭菌方式及残留物、包装材料等。这些是化学表征着重关注的信息,医疗器械材料化学表征的主要对象及风险关注点见表1。③同类产品的历史评价数据、已上市同品种医疗器械的化学表征报告、毒理学数据、临床使用数据、监管部门审批资料等。这些是生物学等同性判定的基础。应当注意到,收集到的信息可能不完整,根据GB/T 16886.18—2022 的指导精神,必要的信息应当在后续的化学表征中补充。

1.2 医疗器械的生物学等同性判定
医疗器械的生物学等同性判定是GB/T 16886.18—2022[2]遵循“最少必要信息”原则、简化评价流程的节点。根据已有资料,若拟用医疗器械与临床已确立的医疗器械在化学特性、物理特性、临床使用及生物学终点等方面足够相似,不会引入额外或不同的毒理学与生物相容性问题,则无需开展额外的化学表征和毒理学风险评估,可直接采信已有生物学安全结论。若根据已有信息,无法给出生物学安全性判定,则需进行化学表征,为后续毒理学风险评估提供数据支撑。
1.3 化学表征
医疗器械的化学表征是一个分层递进的评估过程,旨在全面了解材料的化学特性,主要包含以下4 个部分:医疗器械的组成分析、可浸提物分析、可沥滤物分析,以及材料的结构组成分析。
医疗器械的组成分析是化学表征的第一层次,其目的是在必要的情况下补齐缺失的材料信息。在实践中,企业往往仅能提供材料大类或简单成分说明,难以完整覆盖材料的具体化学成分、添加剂、残留物等信息,也难以准确反映实际生产过程中非有意引入的物质。在必要的情况下,通过系统的成分分析,可以明确医疗器械成分的种类、含量与存在形式,从而为生物学等同性判定、可浸提物和可沥滤物研究、毒理学风险评估等提供必需的数据。
另外,医疗器械成分的最大释放量不会高于该成分在医疗器械中的含量。虽然在临床使用过程中,医疗器械中全部化学成分迁移至人体的情况极为少见,但是通过医疗器械的成分谱,可以确定理论的暴露上限,并给出保守的毒理学风险评估。若该评估结果满足要求,则无需再开展后续的化学表征。
可浸提物分析是医疗器械材料化学表征的第二层次。可浸提物是指, 在严于临床使用的实验室浸提条件下, 从医疗器械中浸提出来的物质。由于根据成分分析无法判断这些物质是否会从材料中释放、是否会进入人体并产生安全风险,因此,需要通过可浸提物分析来筛选出那些可能从医疗器械中迁出的,进而接触人体的成分。“严于临床” 的设计能增加材料中可迁移物质的释放。若所有可浸提物含量均低于毒理学阈值,则可判定为毒理学风险可接受,无需开展后续分析。
可沥滤物分析是医疗器械材料化学表征的第三层次。可沥滤物是指,在临床使用场景下医疗器械释放出来的化学物质。与可浸提物分析采用“严于临床”的实验条件不同,可沥滤物分析的浸提实验需复制或模拟临床使用的场景(在某些情况下,例如对于持久接触医疗器械,可在加速条件下进行模拟浸提)。在这种实验条件下,可沥滤物分析能得到更接近真实情况的析出物质和暴露剂量。可沥滤物分析通常作为可浸提物分析的补充和确认环节。若可浸提物分析发现某类物质的含量接近或超过毒理学阈值时,就需要通过可沥滤物分析进一步验证。需要注意的是,评估人员可以根据实际情况,跳过之前的步骤,直接进行可沥滤物分析。
材料的结构组成分析是医疗器械材料化学表征的重要部分。结构组成分析主要关注材料在分子和超分子层面的组织方式或一些综合指标,如聚合物的构型、结晶性、分子量分布,金属的晶相以及陶瓷的显微结构等。材料的结构组成决定了材料的理化性能(如强度、韧性、弹性),并与材料的降解行为以及长期稳定性相关,因而也是医疗器械生物学评价关注的方面。
在以上化学表征内容中,可浸提物和可沥滤物分析是GB/T16886.18—2022 的核心。可浸提物和可沥滤物分析聚焦“化学物质释放”这一关键点,是衔接医疗器械化学信息与毒理学风险评估的桥梁。有鉴于此,笔者在下文着重介绍可浸提物和可沥滤物分析的技术要点。

02
可浸提物和可沥滤物分析的技术要点
笔者主要围绕样品制备与浸提实验、分析技术选择、定性定量策略及技术环节等,阐述可浸提物和可沥滤物分析方法。
2.1 样品制备与浸提实验
医疗器械的样品制备与浸提实验应按照GB/T 16886.12—2023[4] 的要求规范开展。样品需取自最终产品或采用与最终产品相同工艺制备的材料;如果医疗器械不能整体应用于测试,则选取受试医疗器械的各种材料中有代表性的部分,将其按比例组合成试验样品。样品应根据实际使用形态选择整体浸提或合理裁剪,确保浸提介质能够充分接触医疗器械与人体接触的所有表面。样品制备全过程应避免外来污染,同时保留生产过程中产生的残留物、添加剂、清洗剂等有意或无意引入的成分。如需对样品进行分割,应使用洁净工具,防止工具带来二次污染,并关注切割产生的新表面的影响。
浸提条件应与临床使用场景一致或更严格,以确保获得的可浸提物谱能够覆盖全部潜在可沥滤物,且可沥滤物的释放量不低于临床使用。酸消解等过度剧烈的条件仅适用于成分表征,不可用于可浸提物与可沥滤物分析,以免造成目标物质降解或损失。同时,应避免使用会导致医疗器械明显溶胀或解体的溶剂,这类溶剂会造成浸提液体积测量失真,且医疗器械解体后还可能释放出临床条件下不会释放的物质,影响结果的真实性。
合适的浸提溶剂是实验成功的关键之一[7]。常用的浸提溶剂包括水、生理盐水、乙醇、异丙醇、乙醇- 水混合体系、乙醇- 生理盐水混合体系、人工模拟体液等。依据GB/T 16886.12—2023[4],浸提实验应同时采用极性与非极性两类介质,以保证不同极性的可迁移物质均能被有效提取;仅使用单一浸提介质时,需提供充分的合理性论证。研究表明,乙醇- 水混合体系可作为血液的替代溶剂[8]。针对口腔医疗器械,模拟使用浸提实验中可采用人工唾液[9]。此外,浸提液的pH 同样需要予以关注。浸提液的pH应与临床实际接触环境相近。若医疗器械在临床使用中可能接触不同pH 的液体, 按照GB/T16886.18—2022[2] 的要求, 宜选用2 种不同pH 的浸提介质(如pH=2 和pH=10), 且临床接触液体的pH 范围介于二者之间。
浸提温度与时间的设置需匹配产品临床接触特性。对于长期接触或植入类医疗器械,可设置高于体温的浸提温度(如50℃、70 ℃) 来缩短提取周期, 浸提时间的设定需保证可迁移物质的充分释放。浸提用量( 样品与浸提液的比例)需符合GB/T16886.12—2023[4] 的要求。此外,需同步设置溶剂空白对照, 以排除实验过程中引入的外源杂质干扰。
2.2 分析物
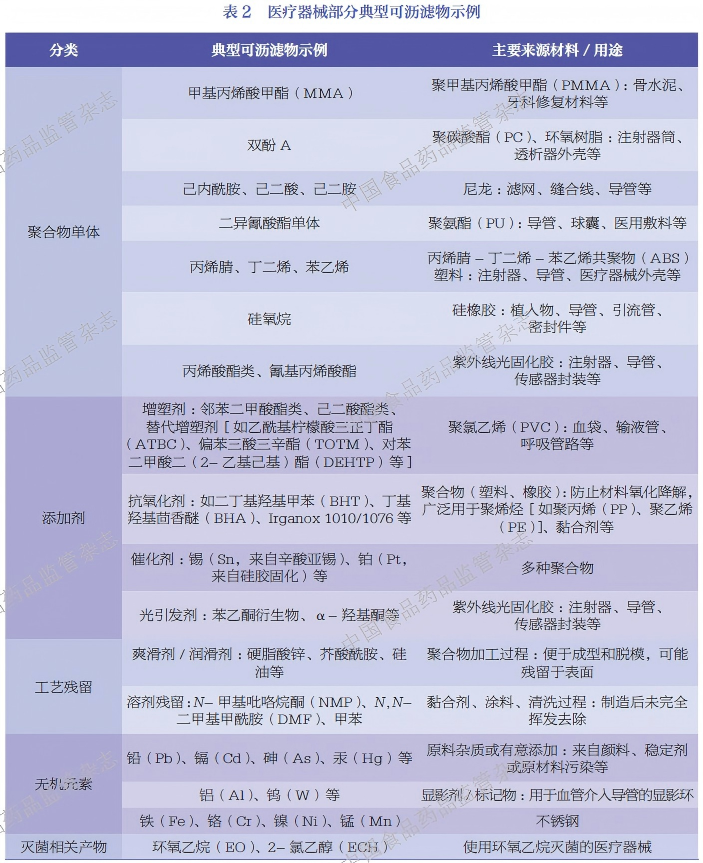
根据识别方式的不同,分析物可分为已知可沥滤物和未知可沥滤物。已知可沥滤物是成分和结构明确的物质,通常可通过医疗器械的材料组成、配方信息、生产工艺、灭菌方式等前期资料确认或预判。除此之外,在可浸提物谱或可沥滤物谱中还存在大量无法预先确定种类与结构的物质,如未知的材料降解产物、副反应产物、加工残留杂质及非预期引入的污染物等,这些物质统称为未知可沥滤物。掌握常见医用材料的典型可沥滤物,有助于快速分析已知可沥滤物,降低鉴定未知可沥滤物的难度,进而更全面、准确地完成医疗器械材料化学表征与毒理学风险评估。医疗器械部分典型可沥滤物见表2。
2.3 分析技术和方法
可浸提物与可沥滤物依据成分类型, 可划分为有机物( 挥发性、半挥发性、非挥发性)、元素及离子三大类, 需采用不同的分析技术检测。针对有机物, 通常采用气相色谱(gas chromatography,GC) 与高效液相色谱(high performance liquid chromatography,HPLC),通过色谱与不同的检测器,或色谱- 质谱联用,实现高效的微量和痕量分析。常用的GC 检测器有氢火焰离子化检测器(flame ionization detector,FID)、电子捕获检测器(electron capture detector,ECD)等。常用的HPLC 检测器有紫外检测器、电雾式检测器(charged aerosol detector,CAD)、蒸发光散射检测器(evaporative light scattering detector,ELSD) 等。核磁共振波谱法(nuclear magnetic resonance spectroscopy,NMRS) 可用于识别 1H 和13C 在分子中的化学环境,解析有机物官能团特征与分子骨架连接关系,实现有机物结构定性,也可以依据峰积分进行定量分析。电感耦合等离子体质谱(inductively coupled plasma mass spectrometry,ICP-MS)、电感耦合等离子体原子发射光谱(inductively coupled plasma atomic emission spectroscopy,ICP-AES)可实现痕量金属元素的精准定量。离子色谱法(ion chromatography,IC) 则可用于分离与检测无机阴离子、金属阳离子、小分子有机酸、小分子有机碱等物质。
医疗器械的化学表征有双重目的:①分析样品中的已知可沥滤物。②筛选样品中的未知可沥滤物,并对超过毒理学阈值的未知可沥滤物进行进一步评估。为此,研究人员分别建立了目标分析方法[10] 与筛选分析方法[11] 两套分析策略。
目标分析方法是针对已知可沥滤物的精准检测方案,其核心是根据目标物质的理化性质(如分子量、极性、挥发性),构建专属的分离、定性与定量流程。该方法依赖标准物质(如纯品或认证参考物质)绘制校准曲线,以确保方法的检出限、定量限、精密度和回收率等参数符合验证要求,从而实现对特定目标物质的准确定量分析,直接为毒理学风险评估提供可靠的暴露数据。
相比之下,筛选分析方法是面向未知可沥滤物的广谱筛查方法,旨在识别可浸提物谱与可沥滤物谱中的潜在高毒理学风险物质。通常采用气相色谱– 质谱联用(gas chromatography–mass spectrometry,GCMS)、液相色谱– 质谱联用(liquid chromatography–mass spectrometry,LC-MS)开展非目标性全扫描检测,通过半定量方法[12],剔除低关注度成分,再对留存的可疑物质开展结构推定与进一步毒理学研判,实现对未知潜在风险的排查。
未知可沥滤物的筛选分析工作需要结合化学分析和毒理学评估, 本质上也是毒理学风险评估的预筛查。分析评价阈值(analytical evaluation threshold,AET) 是筛选未知可沥滤物的有力工具。对于浓度低于AET 且在AET 适用范围内的未知可沥滤物,无需进一步的毒理学风险评估。AET 的计算公式如下。
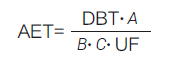
上式中,A 表示用于浸提实验的医疗器械的数量;B 表示浸提液的体积(ml);C 表示临床应用中1 天内与人体接触的医疗器械的数量;DBT( dose-based threshold)表示基于剂量的毒理学阈值(μg/d);UF(uncertainty factor)表示分析方法的不确定因子,例如,在半定量分析中,若采用替代标准品校正会引入误差,此时需根据实际情况调整UF的值[13] ;计算得到的AET 的单位为μg/ml。
DBT 通常采用基于大量物质毒理学数据建立的通用型毒理学阈值, 如毒理学关注阈值(threshold of toxicological concern,TTC)[14]。TTC 等通用型毒理学阈值可为一大类物质设定统一的毒理学阈值,从而极大简化未知可沥滤物的筛选分析工作。但应当指出,TTC 不适用于“关注队列”(cohort of concern,CoC)物质。CoC 物质是一类高毒性物质,包括黄曲霉毒素、N- 亚硝基化合物、偶氮化合物等。TTC 也不适用于高分子聚合物、颗粒物、陶瓷、蛋白质、放射性成分等,因为这些物质并未被纳入用于确立TTC 的数据库中。在应用AET 前,必须通过初步鉴定(如质谱分析)预先排除这些不适用的物质。
此外,需特别强调,AET 原则上仅适用于有机物。这是因为,通用型的毒理学阈值(如TTC)主要建立在有机物的数据基础上,如基于Cramer 结构分类的TTC。对于金属元素,尽管已有关于AET 的探讨[15],但由于金属的毒性具有高度特异性,且受价态、溶解度和生物可利用性等因素影响,难以应用通用型的毒理学阈值。目前,对于金属元素的AET 尚未形成共识。

03
化学表征报告要点
化学表征报告应包括以下内容:①医疗器械基本描述。②样品制备方法。③浸提实验。④所用化学分析技术说明,目标分析方法需附方法验证信息(包括校准曲线、检出限、定量限、精密度和回收率等),以及AET 的计算过程与合理性论证。⑤所有定性、定量及半定量分析结果。
若检测出高毒性物质(如致癌物、内分泌干扰物等)或含量超过AET 的未知可沥滤物,报告中须予以重点说明和详细描述。此外,报告中亦可纳入从其他来源获取的相关化学或组成信息(如材料供应商提供的数据、公开文献资料),但须按上述相同标准要求进行报告,并讨论该信息与毒理学风险评估的相关性。

04
小结与展望
GB/T 16886.18—2022 作为我国医疗器械生物学评价“表征先行”策略的专项标准,构建了从信息收集、生物学等同性判定到多层次化学表征的技术路径,明确了可浸提物与可沥滤物分析的技术规范,为医疗器械材料化学表征提供了统一的实施依据。严谨的化学表征有助于最大限度地减少动物试验,提升生物学评价效率,助力医疗器械的产品设计与风险管理。
立足当前医疗器械行业的发展趋势, 以GB/T 16886.18—2022 为基础的化学表征标准体系将进一步支撑医疗器械行业高质量发展。未来,伴随分析技术的进步、毒理学数据库的完善与标准体系的持续优化,化学表征将在医疗器械全生命周期的风险管理中发挥更关键的作用,为我国医疗器械产业创新与科学监管提供更坚实的技术保障。

第一作者简介
卢忠,研究生,中国药品监督管理研究会医疗器械监管研究专业委员会副主任委员,制药高级工程师。专业方向:医疗器械监管及产业经济研究
通讯作者简介
孔繁圃,硕士,清华大学医学院,研究员。专业方向:医疗器械检测与研究

【参考文献】略




编辑:向丽
审核:赵燕宜






