 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库Cell:代谢分流决定命运,半胱氨酸如何重塑CD8⁺ T细胞功能
发布时间:2026-05-19来源:bioart
CD8⁺ T
细胞是抗肿瘤与抗感染免疫中的核心执行者。在识别抗原后,这类细胞迅速被激活,进入增殖状态,并获得杀伤靶细胞以及分泌干扰素γ
(
IFNγ
)
和肿瘤坏死因子
(
TNF
)
等效应功能。然而,在肿瘤和慢性感染环境中,
T
细胞常因浸润受限、抗原持续刺激或逐渐进入功能耗竭状态,而难以有效清除病灶
【1】
。
近年来的研究逐渐表明,
决定
T
细胞命运的,不仅是抗原与共刺激信号,还包括其能否完成与功能需求相匹配的代谢重编程
【2】
。
氨基酸正是这一过程中不可或缺的关键底物。它们不仅参与蛋白质合成,还深度介入能量代谢、信号转导及氧化还原稳态调控。值得注意的是,一些在稳态条件下可由细胞自身合成的“非必需氨基酸”,在免疫激活等特定情境下也可能转变为限制性因素
【3】
。更为关键的是,氨基酸进入细胞后并不会沿单一路径被利用,而是被分配至多条代谢分支
【4】
。不同代谢通路对细胞功能的支持并不相同,因此,
营养物在细胞内“流向何处”,可能比“是否充足”更加关键
。这一点为理解T细胞功能受限及肿瘤免疫逃逸提供了新的解释框架。
近日,约翰霍普金斯大学
Erika L. Pearce
实验室等在
Cell
杂志发表了题为
Sulfur partitioning from cysteine controls T cell proliferation and effector function
的研究文章,
系统解析了
CD8⁺ T
细胞中半胱氨酸的代谢去向及其功能意义
。
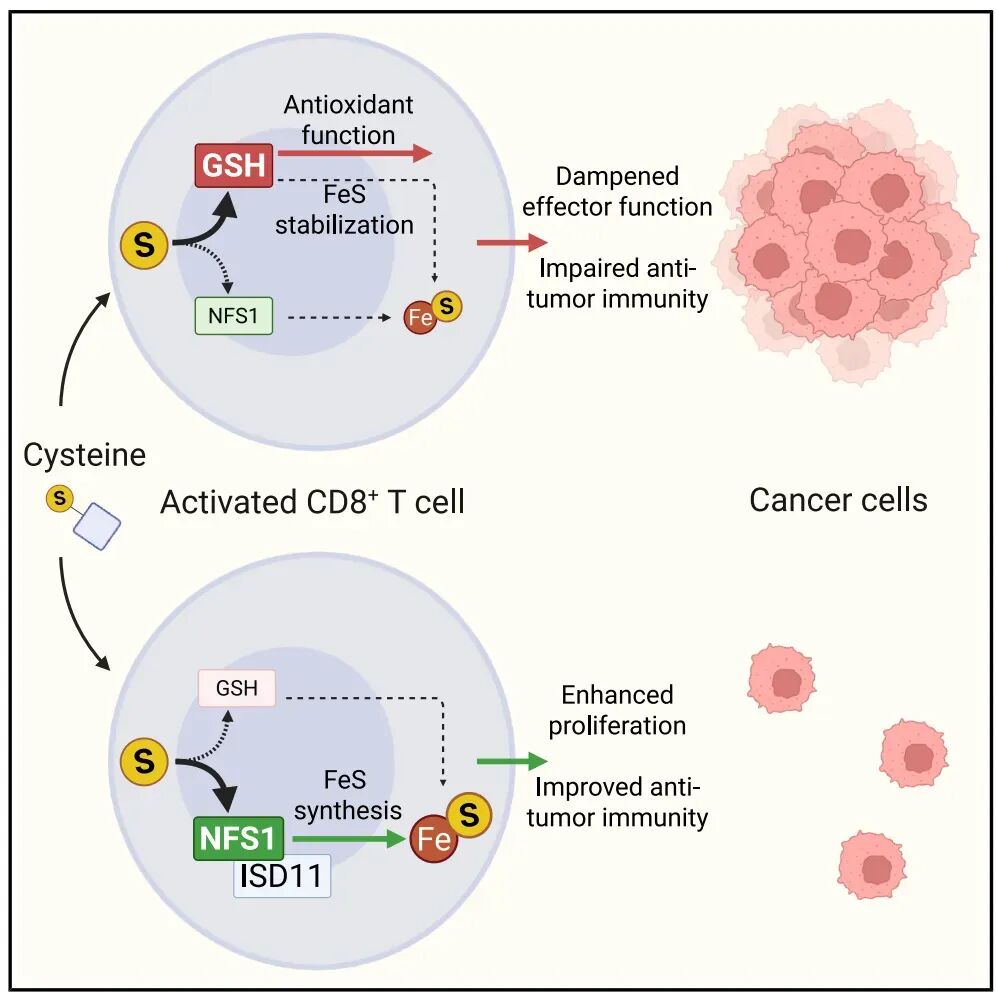
研究发现,摄取的半胱氨酸在细胞内主要分流至两条路径:
一条用于谷胱甘肽(
GSH
)合成,另一条经由
NFS1
进入铁硫簇(
Fe-S
)生物合成
。
这两条路径分别调控
T
细胞的效应功能与增殖能力。
Fe-S
代谢受损会推动
T
细胞耗竭并削弱抗肿瘤免疫,而限制
GSH
通路或增强
Fe-S
代谢则有助于改善肿瘤控制。该研究提出,营养物的功能并不取决于摄入量本身,而取决于其在细胞内的代谢分配。

作者首先将体外激活的效应
CD8⁺ T
细胞置于不同氨基酸缺乏条件下进行分析。结果显示,短期半胱氨酸饥饿虽可显著降低细胞内半胱氨酸水平,但并不影响细胞活力,这与甲硫氨酸饥饿诱导的细胞死亡形成鲜明对比。值得注意的是,半胱氨酸缺乏的
T
细胞呈现出一种特征性状态:
效应功能增强,包括
IFNγ
与
TNF
表达上升以及细胞毒性增强,但增殖能力显著下降,细胞周期停滞于
G0/G1
期
。同时,这种“硫饥饿”状态还抑制了快速增殖细胞的线粒体呼吸,却对静息的记忆
T
细胞影响有限。基于半胱氨酸饥饿产生的“增强效应而抑制增殖”的分离表型,作者进一步追溯了半胱氨酸在细胞内的代谢去向。
通过同位素示踪结合核磁共振与质谱分析,作者发现半胱氨酸被大量用于GSH及其相关代谢物的合成,包括GSSG与半胱氨酰甘氨酸,同时也可进入乙酰辅酶A等代谢网络。GSH合成抑制剂
BSO
能够有效阻断该通路。半胱氨酸缺乏特异性地导致GSH及GSSG耗竭,而其他氨基酸缺乏则不产生类似效应。体内实验同样证实,半胱氨酸是GSH合成的主要来源。进一步分析显示,T细胞激活过程中GSH合成酶表达上调,而还原酶表达下降,提示
激活状态下细胞更依赖从头合成GSH
。
进一步的代谢流分析表明,经典的转硫化途径在激活的
CD8⁺ T
细胞中并不活跃。来自丝氨酸的碳与氮并不会进入半胱氨酸骨架,而仅用于
GSH
中的甘氨酸部分。尽管半胱氨酸缺乏可诱导转硫化酶
CTH
表达上调,但无论是基因敲除还是药理抑制
CTH
,均不影响
GSH
水平及
T
细胞功能。体内实验亦显示
CTH
缺陷对
T
细胞无明显影响。这些结果表明,
激活的
CD8⁺ T
细胞缺乏有效的转硫化能力,因而半胱氨酸在这一状态下表现为条件性必需氨基酸
。
在明确半胱氨酸进入GSH通路后,作者进一步分析该路径对T细胞功能的影响。研究发现,在半胱氨酸充足条件下,GSH对细胞增殖并非必需,但在缺乏半胱氨酸时可部分支持增殖。更为重要的是,抑制GSH合成
(无论通过BSO处理还是GCLC敲除)
均可增强IFNγ表达,提示
持续的GSH生成对效应功能具有抑制作用
。此外,GSH在T细胞中的作用具有时间依赖性:在初始激活阶段,其从头合成对细胞分化至关重要,而在完全激活后,其重要性相对下降。
进一步观察发现,
半胱氨酸缺乏会引发脂质过氧化,而铁螯合剂DFO能够缓解这一现象,提示该过程与细胞内游离铁水平相关
。NFS1作为铁硫簇生物合成的关键酶,可从半胱氨酸中提取硫原子用于Fe-S组装。研究显示,NFS1缺失显著抑制T细胞增殖,导致细胞周期停滞、铁硫簇依赖酶表达下降以及线粒体呼吸受损。同时,NFS1缺陷降低IFNγ产生,却提高TNF表达,呈现与半胱氨酸缺乏不同的功能表型。由于此时半胱氨酸仍可用于GSH合成,细胞内GSH水平反而升高,而抑制GSH仍可恢复IFNγ表达。这表明两条代谢路径在功能上彼此独立,分别调控不同生物学过程。
进一步机制分析发现,半胱氨酸缺乏或
NFS1
缺失均导致游离
Fe²⁺
水平升高,铁硫簇稳定性受损。铁水平升高既来源于
CD71
介导的铁摄入增加,也来源于铁硫簇破坏后的释放。铁螯合并不能恢复增殖能力,说明
铁代谢紊乱是下游结果而非驱动因素
。铁硫簇受损进一步影响
ABCE1
与
DNA2
等关键蛋白,激活
p53
信号并上调
p21
,从而抑制细胞周期进程。
GSH在这一体系中发挥双重作用。一方面,其抗氧化功能可调节IFNγ产生;另一方面,其通过线粒体谷氧还蛋白GLRX5稳定铁硫簇,从而支持细胞增殖。利用牛磺酸或
TEMPO
等抗氧化剂,作者成功区分了这两种功能:抗氧化作用仅影响IFNγ表达,而无法恢复增殖或铁硫簇稳定性。这表明,
GSH的抗氧化功能主要调节IFNγ的产生,而其通过GLRX5稳定铁硫簇的功能则是支持细胞增殖所必需的,两种功能可被功能性地分离
。此外,GSH补充对半胱氨酸饥饿T细胞的保护作用并非通过分解GSH恢复半胱氨酸来实现,而是通过GSH直接稳定铁硫簇的机制发挥作用。
在体内肿瘤模型中,
NFS1
缺陷的
CD8⁺ T
细胞在
B16-OVA
黑色素瘤模型中表现出浸润能力下降、耗竭标志物上调以及抗肿瘤能力减弱。
慢性刺激模型进一步显示,
NFS1
缺失会加速
T
细胞耗竭,并伴随
Fe²⁺
持续升高。相反,
NFS1
过表达或
GCLC
敲除均可增强
T
细胞抗肿瘤功能。对人类肝细胞癌单细胞数据的分析同样支持这一结论:
耗竭
T
细胞中
NFS1
与
FXN
表达降低,而铁摄入相关分子
CD71
上调,提示铁硫代谢失衡与
T
细胞功能衰退密切相关
。
综上所述,本研究揭示了
半胱氨酸在
CD8⁺ T
细胞中的生物学作用,并非由其供给量单独决定,而是取决于其在细胞内被分配至何种代谢通路。
GSH
合成与
NFS1
依赖的铁硫簇生成围绕同一底物形成分流关系,分别调控效应功能与增殖潜能,从而在功能层面构成“效应—扩增”的平衡
。
该研究提出,
免疫代谢调控的关键不在于单纯补充或限制某种营养,而在于精细调控其在不同代谢分支之间的流向
。
这一认识为理解
T
细胞耗竭机制及优化肿瘤免疫治疗策略提供了新的理论基础。
原文链接:
转载说明:本文系转载内容,版权归原作者及原出处所有。转载目的在于传递更多行业信息,文章观点仅代表原作者本人,与本平台立场无关。若涉及作品版权问题,请原作者或相关权利人及时与本平台联系,我们将在第一时间核实后移除相关内容。





