 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库Carl June等发文:CAR-T长期安全性随访或可从15年降至5年
背景
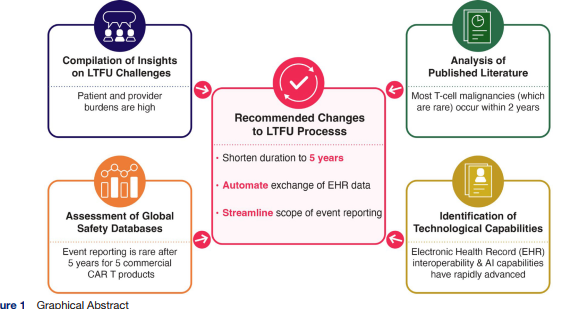
自2017年FDA批准首个CAR-T细胞疗法以来,其长期安全性曾属未知;如今随着超3万患者接受治疗及7款产品获批,长达15年的临床数据已充分证实该疗法具有积极的获益风险比及潜在治愈力。鉴于当前良好的临床应用成果与技术环境的成熟,学界呼吁重新审视既有的长期随访(LTFU)监管流程与要求,以确保其兼具科学依据且真正以患者为中心。
2026年4月,来自美国Catalyst医疗咨询公司的Betsy Foss-Campbell等在《Journal for ImmunoTherapy of Cancer》上发表了题为“Data-informed optimization of CAR T-cell therapy long-term follow-up”的文章。为重新评估15年随访期的科学依据,研究者召集了由患者权益组织、学术界、产业界及政府代表组成的多方利益相关者工作组,宾夕法尼亚大学Carl June教授也是署名作者之一,文章建议基于累积的大量安全性数据,现有的长期随访要求应当被简化和缩短,在临床试验及商业化场景中,5年随访期已具备充分科学合理性。此外,提议利用技术进步,简化流程,将电子健康记录中的重点安全数据自动导入第三方数据库。为推动落地,建议使用成熟平台对更新的数据收集方案进行可行性测试,并阐述了促进采纳这些建议的监管政策考量。
研究结果
LTFU数据收集要求和流程
目前的CAR-T疗法利用整合型病毒载体将CAR基因导入T细胞,因存在插入性致癌的理论风险,而引发了对第二原发恶性肿瘤(SPM)的担忧,导致FDA迄今要求所有临床试验受试者及部分商业患者接受长达15年的长期随访数据收集,并在2023年11月进一步强调需对患者实施终身监测。如下文详述,现行的长期随访数据收集要求与流程极为繁重复杂。
临床研究管理
根据FDA指南,临床试验中的CAR-T疗法受试者需建立完整的病例档案,并在输注后5年内定期接受医护人员体检,随后10年每年至少随访一次。研究者负责收集长期随访数据并提交给申办方,再由申办方上报至FDA(见图1)。
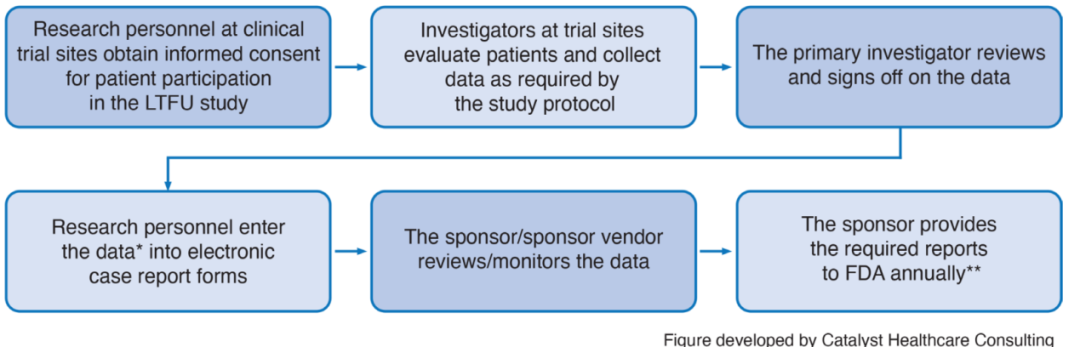
图1 临床试验受试者长期随访(LTFU)数据采集的典型流程
商业化CAR-T产品的管理
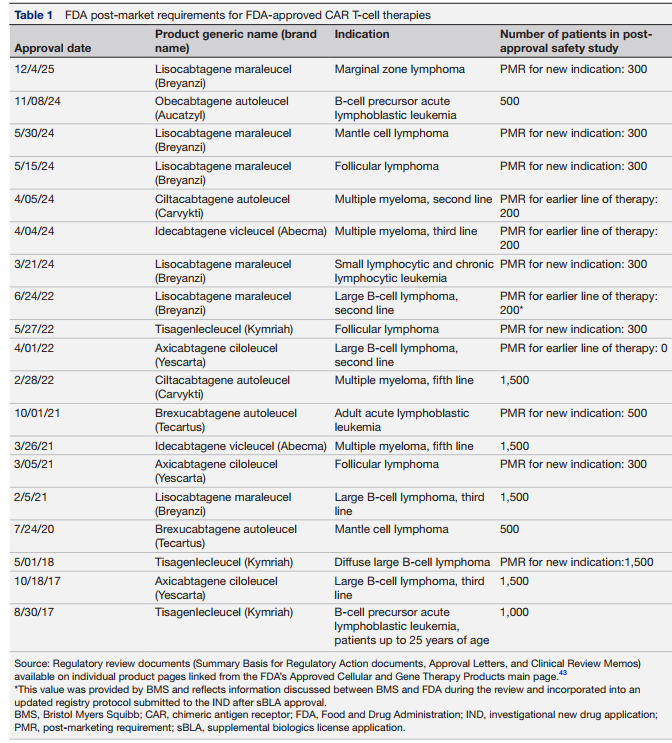
在商业化应用中,FDA通过针对各产品的上市后要求(PMR)实施安全监管,目前已获批的CAR-T疗法均须开展长达15年的随访研究,初始适应症需追踪多达1500名患者,新增适应症则需额外纳入300至1500名患者,仅针对早期治疗线数的产品可适当缩减样本量(如表1所示)。此类上市后安全性研究的数据收集同样繁重,治疗中心通常需向国际血液与骨髓移植研究中心(CIBMTR)运营的主注册库报送数据。数据管理员必须人工翻阅患者电子病历,填写总计约14份、单份含7至522个问题的表格,涵盖从基线到每年随访的所有节点,其中多数表格适用于所有受试者。图1直观展示了这一典型的上市后注册研究的全流程。
表1 FDA批准的CAR-T细胞疗法上市后的要求
延长安全性监测
CAR-T疗法的延长安全性监测包含监管要求的15年随访与上市后研究,以及2023年11月FDA安全通讯提出的终身监测建议,后者要求对接受靶向B细胞成熟抗原或CD19自体CAR-T治疗的患者终身监测继发性恶性肿瘤,通过标准医疗护理实现,医疗机构需向生产商报告事件并指导采集患者样本检测CAR转基因,同时将疑似不良事件上报FDA的MedWatch项目,相关数据存入FDA不良事件监测系统数据库。此举源于治疗后罕见的继发性T细胞恶性肿瘤病例报告,尽管个别案例检出CAR转基因,但尚未证实其与T细胞恶性肿瘤的因果关系,且患者常因既往化疗等治疗存在T细胞恶性肿瘤背景风险升高因素。值得注意的是,2024年11月,时任FDA生物制品评价与研究中心主任的Peter Marks公开指出,在发布安全通报时,并未正式考虑这一背景事件。
当前LTFU数据收集要求和流程的挑战
当前长期随访流程冗长且复杂,极大增加了患者及医疗机构的负担。具体如下:
患者负担与失访
艾米丽·怀特黑德基金会与 Catalyst 医疗咨询公司近期联合开展的一项研究显示,在对近 100 名 CAR-T 疗法受试者进行随访时发现显著的患者流失现象。值得注意的是,在完成输注已超过一年的受试者中,有 20% 已不再坚持随访就诊。此外,近期(一年内)接受过治疗的受访者中,38%的受访者认为自己无法坚持完成 15 年的随访,其中大多数人认为自己最多只能坚持8年。调查指出,患者参加随访面临的最大挑战均与出行相关,包括往返治疗中心的路程遥远及交通成本高昂。
供应商或研究者负担:耗时和成本
无论临床试验还是商业化应用,CAR-T疗法的长期随访(LTFU)流程均步骤繁杂且资源消耗巨大。研究发起者往往缺乏专项经费,迫使学术医疗中心自费雇佣专员进行繁琐的手工数据录入,而在商业场景中,单个中心甚至需配置多达6名专职人员。然而,高昂的人力成本远超过登记系统给予的补偿,且当患者在社区复诊时,中心难以跨机构调取数据。随着CAR-T疗法向社区推广,本就缺乏资金和时间的社区肿瘤科医生更难承受此重负,这迫切要求简化数据收集流程以适应新的医疗场景。
应对挑战的解决方案
根据证据重新评估随访时间
FDA关于基因治疗产品给药后长期随访(LTFU)的指南指出,LTFU的目标是“识别并降低接受[基因治疗]产品患者的长期风险”。为契合这一目标,我们在评估整合型载体的适宜随访时长时,采用了基于风险的考量方式。相应地,我们的评估聚焦于临床与机制层面最相关的不良事件(AE)。具体而言,我们通过对比CAR-T疗法与标准治疗(SOC)后第二原发恶性肿瘤(SPM)的发生率,评估插入突变的理论风险是否已成现实,并探究其与产品的因果关系。如下文详述,分析显示:不良事件通常在3年内报告,5年后罕见;绝大多数SPM发生于5年内;T细胞恶性肿瘤尤多在输注后2年内报告。此外,尚无证据表明CAR-T疗法与SPM(含T细胞恶性肿瘤)间存在经插入致癌作用导致的直接因果关联。
5款CAR-T产品治疗后AE报告汇总
为评估CAR-T疗法后不良事件的发生时间,本研究汇总了5款FDA批准产品的关键临床试验数据。鉴于临床试验中的长期安全性报告较上市后监测更为系统完整,开发者对其全球安全数据库进行了全面检索,以分析不良事件在各随访年份的报告比例(图2)。数据显示,在中位随访24至64个月期间,大多数不良事件(53%–88.6%)集中在输注后第一年报告;此后占比急剧下降,第二年5.5%–18%,第三年3.1%–24%,第四年0.6%–6.5%,第五年0%–8.5%;五年后更是降至极低水平(第六年0%–2%,第七年0%–0.2%)。这表明不良事件主要在三年内报告,五年后极为罕见。五年后持续报告的少数事件多为感染和继发恶性肿瘤,这与接受CAR-T治疗的免疫抑制人群预期相符,且与标准治疗后的背景风险一致。需注意的是,受限于不同数据库的收集差异、随访时长不一及患者流失等因素,这些数据的可比性存在一定局限。
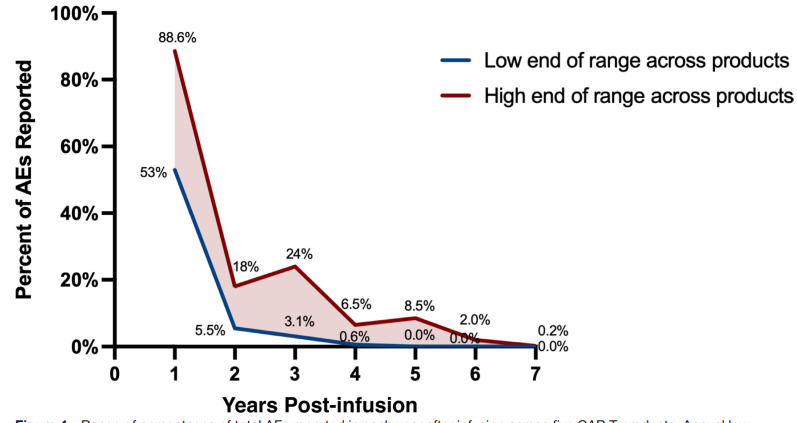
图2 五种CAR - T产品输注后每年报告的总AE百分比范围。
文献证据:CAR-T继发恶性肿瘤风险与标准治疗相当,长期随访可缩至5年
鉴于化疗和/或放疗可能诱发继发性原发性恶性肿瘤(SPM),本研究基于相关文献对比了CAR-T疗法与标准治疗(SOC)的SPM发生风险。数据显示,在最长6年的随访期内,CAR-T疗法后的SPM发生率(0.4%–6.2%)与SOC治疗(4%–6.7%)相当(表2)。在5年随访节点,混合队列中CAR-T治疗后的SPM累积发病率为2.8%(95% CI: 1.7%–4.5%)。作为参照,成人多发性骨髓瘤自体移植后6年的SPM发生率为5.3%。值得注意的是,虽然总体发生率低,但血液系统恶性肿瘤(如骨髓增生异常综合征、急性髓系白血病)在CAR-T治疗后的发生率(0.8%–2.2%)略高于SOC组(0%–0.9%)(表3)。
表2 CAR-T和标准疗法发生SPM几率的对比
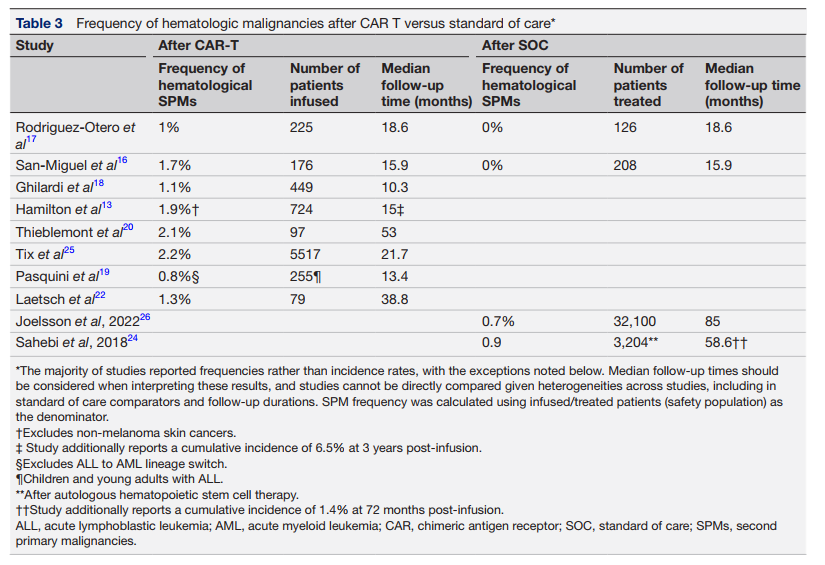
虽然血液系统恶性肿瘤在CAR-T治疗后略高(0.8%–2.2% vs. SOC 0%–0.9%)(见表3),但这主要归因于患者因素:CAR-T受众通常为高龄且历经多线治疗的复发/难治群体,其本身即是高危人群。例如,原发性纵隔B细胞淋巴瘤患者常接受R-CHOP或DA-EPOCH-R等强化疗。Gazeau及其同事在对2017年9月至2023年8月期间接受CAR-T细胞治疗的511例非霍奇金淋巴瘤患者进行的回顾性分析中报告,患者年龄和既往治疗线数是发生治疗相关髓系肿瘤(如急性髓系白血病和骨髓增生异常综合征)的统计学显著风险因素。在另一份近期报告中,Farina及其同事指出,大多数在CAR-T细胞治疗后发生继发性髓系恶性肿瘤的患者表现出高风险突变(del7和TP53)。部分患者在治疗前即存在基因改变,表明这也是SPM发生的不良预后因素之一。
表3 CAR-T和标准疗法发生血液恶性肿瘤几率的对比
由于CAR-T技术涉及基因整合,T细胞恶性肿瘤是关注焦点。值得注意的是,美国FDA官员去年得出结论,CAR-T细胞疗法后的T细胞恶性肿瘤非常罕见。FDA报告称,截至2023年底,其已获悉在使用当时获批的6种疗法中的5种进行治疗后出现了22例T细胞恶性肿瘤病例,但由于样本量较小且CAR-T细胞产品存在差异,尚无法就其与治疗的潜在关联得出结论。重要的是,FDA官员表示:“在美国已施用超过27000剂六种获批产品的情况下,即使假设所有报告的病例都与治疗相关,接受CAR-T细胞疗法的人群中T细胞癌症的总体发生率似乎也非常低。” 已报道CAR-T细胞疗法后罕见T细胞恶性肿瘤的研究显示,其发生率远低于1%(0.09%–0.5%)。在FDA的报告中,3例进行了基因测序的T细胞恶性肿瘤病例均显示出CAR转基因的存在。然而,肿瘤性CAR阳性T细胞的存在本身并不能证明CAR是导致恶性肿瘤的原因,且CAR-T细胞在T细胞恶性肿瘤发生中的因果作用尚未得到证实。
更重要的是,CAR-T治疗后新发恶性肿瘤多较早出现,2025年研究显示783名受试者中除1例有颈部放疗史者第14年患甲状腺癌外,绝大多数在输注后5年内发病,22%随访超5年;420名CD19 CAR-T患者中,继发性病变中位确诊3.2年,5年累积发病率1.5%;CIBMTR数据显示11345名商业CAR-T受体中,输注至首次病变中位时间9个月;FDA报告14例T细胞恶性肿瘤均在给药后2年内发生。结合FDA基于风险调整监管的历史先例,建议将长期随访(LTFU)数据收集期限缩短至5年。
基于风险方法的管理先例
FDA曾依据新数据调整长期随访要求:2006年因潜在风险建议AAV载体随访15年,2020年基于其良好安全性数据,将AAV载体随访期缩短至5年。两份指南均强调应随技术和数据积累重新评估随访建议,体现了FDA基于风险证据灵活调整监管策略的一贯立场。
使过程现代化:数据收集的技术工具和标准
研究者针对CAR-T细胞疗法等生物制品的上市后长期随访数据收集提出了系统性改革方案。现行流程高度依赖人工填报,不仅给医疗机构和制药企业带来了沉重的行政负担,且数据捕获效率低下。尽管监管机构已着手缩短随访时长要求,但数据采集过程的繁琐性仍是核心痛点。
本文主张构建一个去中心化的自动化数据交换体系作为长期解决方案。具体而言,建议建立与FDA合作的第三方数据库,直接对接各大医疗机构的电子健康记录系统,实时获取去标识化的真实世界数据。这种“直通式”传输不仅能省去多重人工录入环节,还能使患者在当地接受随访时就同步完成数据抓取,从根本上提升监测效率并降低各方负担。
这一愿景正逐步具备落地基础。技术层面,EHR系统的互操作性已取得显著突破,美国已建立起全国性的健康信息共享框架,主流EHR供应商及各类技术标准正逐步实现数据格式的规范化。监管层面,FDA的“生物制品有效性与安全性倡议”已通过试点项目验证了可行性:该系统能利用人工智能自动检测不良事件,并从多家医疗机构成功获取符合监管要求的临床数据,且整个过程严格遵循隐私法规,确保原始数据不出医院防火墙,仅传输去标识化信息。
基于此,文章提出具体建议:短期内应利用现有的BEST平台,优先针对CAR-T疗法开展自动化数据交换的可行性评估;长期来看,应将此模式推广至集中式数据库管理。同时,建议优化数据采集策略,摒弃“广撒网”式的报告,转而采用更精确的医学定义,将随访重点聚焦于与CAR-T疗法高度相关的特异性不良事件。通过打通医疗系统与监管系统的数据壁垒,以技术赋能替代低效的人工操作,最终构建起一个高效、精准且低负担的下一代药物安全监测体系。
研究者建议
第一,将CAR-T细胞临床试验及已上市产品的批准后安全性研究的长期随访要求缩短至5年。具体建议FDA修订相关指南,明确对于使用整合载体的CAR-T产品,推荐的延迟性不良事件监测时长通常为5年,并将此标准纳入即将发布的细胞与基因治疗(CGT)上市后数据捕获指南终稿中。同时,鉴于上市后数据可与临床试验数据互为补充,建议酌情缩减上市后要求(PMR)的受试患者数量。此外,除主动监测外,对于临床试验及商业化用药患者,可继续通过“MedWatch”系统维持医护人员的自发被动监测,以实现科学监管与资源优化的平衡。
第二,通过实现电子健康记录数据与中央第三方数据库的自动化交换,简化不良事件数据收集流程。 建议长远开发类似FDA“BEST计划”的平台,依托与FDA合作的第三方数据库,实现医患数据的安全自动化传输。建议CBER的BEST团队先行评估该平台在CAR-T及基因治疗领域的可行性。鉴于现行手工上报负担沉重,建议FDA更新指南,删除强制要求建立或利用登记处的规定,赋予申办方灵活选择数据收集方式的权利(如第三方数据库、AI等),作为过渡方案,以降低资源消耗并简化流程。这一建议将使上市后安全性数据的收集流程简化为以下步骤:
研究中心获取患者知情同意; 医护人员对患者进行评估; 医护人员将结果记录在电子健康记录中; 数据从电子健康记录自动传输至中央第三方数据库; 申办方请求并从第三方数据库获取数据,以完成并提交为期5年的FDA报告。
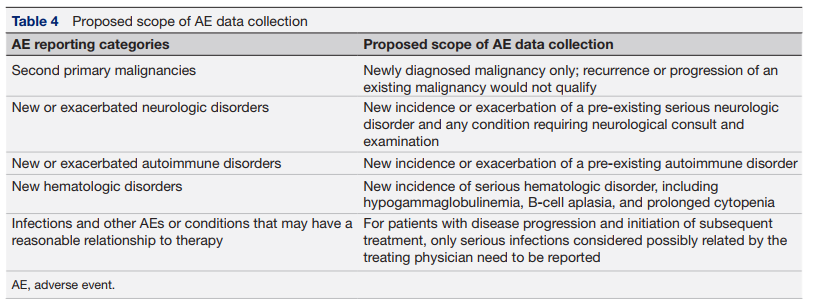
第三,精简不良事件数据收集要求。 建议精简CAR-T疗法长期毒性报告范围,聚焦继发性恶性肿瘤、特定神经及自身免疫疾病、血液系统疾病与感染等重点类别,剔除模糊的关联性事件。通过采用更精确的AE定义(详见表4),可在确保捕获关键安全信号的同时,大幅降低报告负担。建议监管机构与业界协作,删减数据表格中冗余问题,实现更聚焦、高效的上市后监测。
表4 AE数据收集范围的建议
自CAR-T首批临床试验启动至今已有15年,期间已有逾3万名患者接受治疗。事实证明,CAR-T细胞疗法是一种有效、能挽救生命且具有治愈潜力的治疗手段,其安全性特征也已明确。鉴于本文综述的长期安全性数据,目前强制要求的15年长期随访已无必要。优化流程并缩短随访时长,既能保留适当的安全监测,又能最大限度减轻患者、临床医生、申办方及监管机构的不必要负担。
参考文献:
[1] Foss-Campbell, Betsy et al. “Data-informed optimization of CAR T-cell therapy long-term follow-up.” Journal for immunotherapy of cancer vol. 14,4 e013878. 13 Apr. 2026, doi:10.1136/jitc-2025-013878
[2] K Kobbe, Guido et al. “Aggressive Lymphoma after CD19 CAR T-Cell Therapy.” The New England journal of medicine vol. 391,13 (2024): 1217-1226. doi:10.1056/NEJMoa2402730
[3] Hamilton, Mark P et al. “Risk of Second Tumors and T-Cell Lymphoma after CAR T-Cell Therapy.” The New England journal of medicine vol. 390,22 (2024): 2047-2060. doi:10.1056/NEJMoa2401361
[4] Verdun, Nicole, and Peter Marks. “Secondary Cancers after Chimeric Antigen Receptor T-Cell Therapy.” The New England journal of medicine vol. 390,7 (2024): 584-586. doi:10.1056/NEJMp2400209

扫描二维码进群获取最新资讯
关注博生吉细胞研究,获取免疫细胞治疗最新资讯。





