创新药患上了油腻病
发布时间:2026-06-05来源:药事纵横
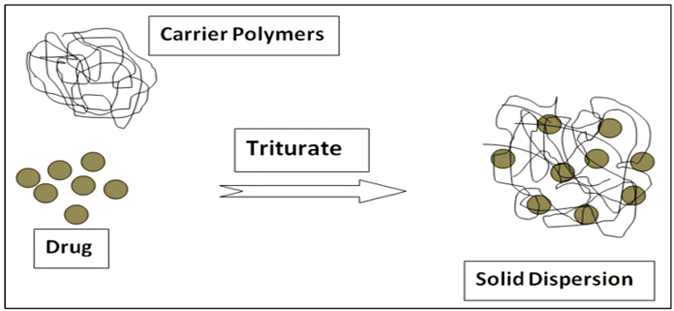
大约在十几年前,我第一次在课上读到Lipinski的五规则。老师把四条标准写在黑板上,分子量不超过500,logP不超过5,氢键供体不超过5,受体不超过10,然后说了一句让我记到现在的话:这可能是药物化学史上最有影响力的四条不等式,但它真正告诉我们的不是怎么设计好药,而是我们这些年做出来的分子,越来越不像药了。当时我并不完全理解这句话的沉重。直到参加工作以后,在项目会上一次次看到优秀的先导因为生物利用度不足5%被叫停,或许需要制剂用喷雾干燥把那些熔点在250℃以上的分子硬生生做成无定形固体分散体(见图1)。后来,我才渐渐意识到,这位老师说的不是一句俏皮话,而是对制药行业过去三十年最精准的预判。图1固体分散体示意图[1]本文试图回答一个问题:为什么小分子创新药越来越难溶解?这不是一个简单的物理化学问题。它背后牵扯着药物化学的优化惯性、组合化学的结构遗产、靶标生物学的演变、以及一种深嵌在制药文化中的“效力至上”偏误。要讲清楚这件事,得从头说起。1.1难溶已经成为“新常态”
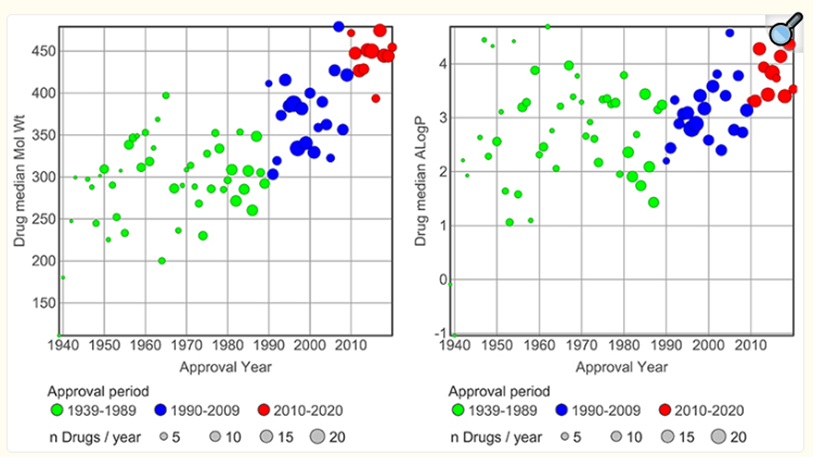
最常被引用的是这个:当前新发现的活性分子中,大约40%到50%在水中的溶解度低到近乎于零。对于制剂科学家来说,这些化合物属于“砖粉”(brick dust)级别。但如果把统计口径缩小到正在被认真推进开发的候选药物,这个比例会跳到70%到90%。也就是说,今天的药物管线中,十个候选物里有七八个从最根本的意义上就无法在胃肠道里有效溶出。即便是已经获批上市的口服药,情况也不乐观。2024年的一项综合分析显示,美国市场上销量前200的口服药中,约40%属于BCS II类或IV类。你可能觉得40%还好,但值得记住的是,这200个药里有很多是二三十年前获批的老品种。如果我们只看2015年以后获批的创新药,这个比例还要高得多。图2本文选取了三个药物审批时间段,依据的是对643种药物(每种药物在其靶点上有至少100个对照化合物与之对比)的分子量和亲脂性(ALogP)增长趋势。三个审批时间段之间的分子量和ALogP差异具有统计学显著性(t检验,p < 0.05)[7]。把时间线拉长,趋势更加触目。1980年代上市的口服小分子药物,中位分子量大概在330道尔顿左右,中位logP约2.5。到了2000年代中期,这两个中位值分别涨到了430和3.5。到了2020年代,虽然涨速放缓,但方向依然明确地向更大、更油的一端移动。2021年Leeson团队对ChEMBL数据库中643个上市药物的全面分析表明,2010到2020年获批的分子,在分子量和亲脂性上与靶标配体化合物已经不存在统计学差异(见图2),这在二十年前是不可想象的。药物曾经因为比配体更苗条而存活下来,如今这个优势正在消失。这些数字背后是一个令人不安的事实:药物化学家们花了三十年在越来越糟糕的化学空间里找分子,而且找到的分子还在继续变糟。1.2溶解度为什么重要
溶解度在药物研发的优先级排序中,地位一直很尴尬。它太基础了,基础到容易被认为是谁都懂的常识,因而在项目会上几乎不会被认真讨论。效力(potency)才是有“新闻价值”的参数:一个IC₅₀从100nM优化到5nM的故事,可以在组会上赢得掌声。而我们把平衡溶解度从2 μg/mL提到了5 μg/mL。即便这在生物药剂学的意义上可能是更重要的进展,可是呢这听起来就是没有那种“势能”。但物理不会因为你忽视它而不起作用。理解溶解度的困境,需要从三个层面来看。第一个层面是热力学。一个结晶药物分子的平衡溶解度,本质上是一个能量博弈:是分子-分子之间的晶格能赢了,还是分子-水之间的溶剂化能赢了。这个博弈的结果浓缩在Yalkowsky的通用溶解度方程里:logS≈0.5−0.01(MP−25)−logP。熔点和logP每升高一点,溶解度就往下走一点。对于那些熔点超过200°C且logP超过4的分子,这在当代药物管线中一抓一大把。方程的预测是残酷的:它们的固有水溶性往往低于1μg/mL。这个量级意味着,即便给足时间,能在250 mL水中(模拟空腹胃液体积)充分溶解的药量,也远远小于产生药理效应所需的最低剂量。第二个层面是动力学。即便平衡溶解度勉强可以接受,还存在一个问题:药能溶多快?Noyes-Whitney方程告诉我们,溶解速率正比于固液界面的面积和浓度梯度。在肠道那短短三四个小时的有效吸收窗口内,一个溶解速率缓慢的分子可能还来不及充分溶出就被排空了。这是溶出限速吸收(dissolution rate-limited absorption)的本质,不是溶不了,是来不及溶。随着分子量增加、扩散系数下降,这个问题只会越来越严重。第三个层面,也是最容易被误解的一个,是过饱和的“虚假承诺”。现代制剂技术,特别是无定形固体分散体(ASD),能够将分子强行推入一种高能的非晶态,使表观溶解度达到晶型平衡值的数百倍。这是在体外呈现的结果。但过饱和是热力学亚稳态的。被你用聚合物强行按住的无定形分子,无时无刻不在寻找回到晶体最低能态的路。肠道环境的复杂性,如pH在胃和十二指肠之间剧烈跳变,胆汁盐和磷脂形成的混合胶束既是增溶帮手又可能是成核催化剂,食物成分的介入让介质的组成变得不可预测,这些使得体内的过饱和维持时间和程度几乎不可能从体外数据直接外推。更糟糕的是,一旦发生沉淀,形成的往往不是原来的晶型,而是一种更稳定、更不溶的形态。你不仅浪费了制剂,还让问题变得更难收拾。Fine-Shamir和Dahan在2023年的一篇综述里把这些微妙的关系提炼成了一个清晰的概念:溶解度-渗透性互偿(solubility-permeability interplay)。他们的核心论点是:制剂提高了表观溶解度的同时,往往降低了游离药物浓度和有效渗透性。加更多的表面活性剂让溶出曲线更漂亮,但药物被胶束包裹得越紧,能穿过肠壁的游离分子就越少。他们把最常见的状况称作“tradeoff模式”,即溶解度上去了,吸收率下来了,一加一减,净收益接近于零。这个洞察直指一个被广泛忽视的盲区:只看溶出数据来评价制剂是不够的,你得同时看渗透性。但你如果在发现阶段根本不测渗透性,这是大多数项目的常态,你就永远不知道你的增溶策略到底是在帮忙还是在帮倒忙。1.3“越做越难溶”是一个悖论,而不是一个意外
读到这里,你可能会有一个直觉反应:既然溶解度这么重要,既然三十年前Lipinski就敲响了警钟,既然每个药物化学家在本科阶段就学过“相似相溶”,那为什么分子还是越来越难溶?如果难溶趋势只发生在某个公司、某类靶标、或某个特定的化学策略中,那它可能只是个可以修正的技术偏差。但它不是。GSK看过自己的管线,Pfizer看过自己的,Leeson用ChEMBL公共数据独立验证过,AstraZeneca内部的化合物质量评估体系也没能逆转趋势,不同公司、不同靶标、不同研发哲学,终点却出奇地一致。这暗示着一个更深层的可能性:不是我们不会做可溶的分子,而是我们身处的整个药物发现体系,从激励机制到合成基础设施,从靶标选择的演化逻辑到化合物库的构成,系统性地指向了“更难溶”的方向。药物化学家不是在主动选择做难溶的分子,而是在一张有坡度的台面上行走。你可以挣扎着往坡上爬两步,但只要稍微松懈,就会滑回那个“更大、更油、更难溶”的平衡态。在接下来的文章中,我将追溯这张“倾斜的台面”是如何被构建起来的。哪些力量在把分子推向下坡,我们发展出了哪些理论和工具来对抗这种下滑,制剂科学在多大程度上能兜住这个底,以及最终,有没有可能从根源上改变这种系统性偏误。答案可能比“改改规则就行”要复杂得多。但理解问题的全貌,至少是解决问题的第一步。[1] Mandhare PM, Ghuse HA. Formulation-Driven Strategies for Overcoming Solubility Barriers in Drug Development: A Review. IJISRT, 2026.[2] Khan KU, Minhas MU, Badshah SF, et al. Overview of nanoparticulate strategies for solubility enhancement of poorly soluble drugs. Life Sciences, 2022, 291: 120301.[3] Nyamba I, Sombié CB, Yabré M, et al. Pharmaceutical approaches for enhancing solubility and oral bioavailability of poorly soluble drugs. European Journal of Pharmaceutics and Biopharmaceutics, 2024, 204: 114513.[4] Fine-Shamir N, Dahan A. Solubility-enabling formulations for oral delivery of lipophilic drugs: considering the solubility-permeability interplay. Expert Opinion on Drug Delivery, 2023, 21(1): 111–126.[5] Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. Journal of Pharmacological and Toxicological Methods, 2000, 44(1): 235–249.[6] Wenlock MC, Austin RP, Barton P, Davis AM, Leeson PD. A comparison of physiochemical property profiles of development and marketed oral drugs. Journal of Medicinal Chemistry, 2003, 46(7): 1250–1256.[7] Leeson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nature Reviews Drug Discovery, 2007, 6(11): 881–890.[8] Leeson PD, Bento AP, Gaulton A, et al. Target-based evaluation of "drug-like" properties and ligand efficiencies. Journal of Medicinal Chemistry, 2021, 64(11): 7009–7029.
转载说明:本文系转载内容,版权归原作者及原出处所有。转载目的在于传递更多行业信息,文章观点仅代表原作者本人,与本平台立场无关。若涉及作品版权问题,请原作者或相关权利人及时与本平台联系,我们将在第一时间核实后移除相关内容。
 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库