 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库周末文摘 | 重组蛋白药物研发50年回顾与展望(1976~2026)


引用本文
罗永章.重组蛋白药物研发50年回顾与展望(1976~2026)[J].中国食品药品监管,2026(5):4-37.
重组蛋白药物研发50年回顾与展望(1976~2026)
50 Years of Recombinant Protein Drug R&D:
Retrospect and Prospect (1976~2026)
罗永章
清华大学生命科学学院
蛋白质技术国家工程研究中心
LUO Yong-zhang
School of Life Sciences, Tsinghua University
The National Engineering Research Center for Protein Technology
摘 要 / Abstract
本综述回顾了自20 世纪70 年代重组脱氧核糖核酸技术问世以来,重组蛋白药物五十载的研发历程。文章梳理了以中心法则理论为基础,重组人胰岛素、重组人促红细胞生成素、单克隆抗体等里程碑药物的研发演进轨迹;概述了主要表达系统、高密度发酵和纯化工艺等共性技术。鉴于正确折叠(复性)是重组蛋白药物研发的最大难点之一,本文追溯了蛋白折叠领域的发展历程,重点归纳了折叠中间体理论,尤其是二硫键在折叠动力学中的双重作用:既能锁定蛋白的天然态,也能诱导折叠中间体陷入动力学陷阱,为工业化复性重组蛋白药物(如重组人血管内皮抑制素)提供了理论依据。笔者利用蛋白质折叠理论进一步解读了人血白蛋白结构(17 对二硫键,1 个具有催化能力的自由巯基,及氨基酸残基组成)与功能之间的关系,并归纳提出了“白蛋白688 原理”,即白蛋白占人体血浆总蛋白约60%,提供约80% 的自由巯基并维持约80% 的胶体渗透压,揭示了人类生命系统中白蛋白的浓度和质量作为核心稳态调控子的定量基准;进一步论述了“年轻、无损、超纯”重组白蛋白可能对抗机体衰老,为干预系统性疾病与退行性病变提供重要切入点。据此,笔者建议从蛋白折叠与复杂系统科学等维度进一步丰富重组蛋白药物质量监管的策略,指出在利用硅基智能加速重组蛋白药物成功研发的同时,仍须严守碳基生命的底层法则。
This review looks back on the fifty-year research and development history of recombinant protein drugs since the advent of recombinant DNA technology in the 1970s. The article outlines the research and development evolution trajectory of milestone drugs such as recombinant human insulin, recombinant erythropoietin, and monoclonal antibodies based on the central dogma; it also summarizes common technologies including major expression systems, high-density fermentation, and purification processes. Given that correct folding (renaturation) is one of the greatest challenges in the research and development of recombinant protein drugs, this paper traces the developmental history of the protein folding field and focuses on summarizing the theory of folding intermediates, particularly the dual role of disulfide bonds in folding kinetics: they can not only lock the native state of proteins but also induce folding intermediates to fall into kinetic traps, which provides a theoretical basis for the industrial renaturation of recombinant protein drugs (such as recombinant human endostatin). The author utilizes protein folding theory to further interpret the relationship between the structure of human serum albumin (17 pairs of disulfide bonds, one catalytically active free sulfhydryl group, and its amino acid residue composition) and its function, and inductively proposes the “Albumin 688 Principle”: albumin accounts for approximately 60% of the total protein in human plasma, provides about 80% of the free sulfhydryl groups, and maintains roughly 80% of the colloid osmotic pressure. This reveals the quantitative benchmark of albumin concentration and quality as a core homeostatic regulator in the human life system; furthermore, it discusses how young, undamaged, and ultra-pure recombinant albumin may combat organismal aging, providing an important entry point for intervening in systemic diseases and degenerative pathologies. Accordingly, the author suggests further enriching the quality regulation strategies for recombinant protein drugs from the dimensions of protein folding and complex systems science, pointing out that while utilizing silicon-based intelligence to accelerate the successful research and development of recombinant protein drugs, the underlying laws of carbon-based life must still be strictly respected.
关 键 词 / Key words
重组蛋白药物;蛋白质折叠机制;重组蛋白复性;白蛋白688 原理;监管科学
recombinant protein drug; protein folding mechanism; recombinant protein renaturation; albumin 688 principle; regulatory science
01
重组蛋白药物研发概述(Overview of Recombinant Protein Drug R&D)
自20 世纪70 年代重组脱氧核糖核酸(deoxyribonucleic acid,DNA)技术问世以来,医药产业经历了从“化学小分子”向“生物大分子”的深刻变革。半个世纪间,重组蛋白药物不仅重塑了肿瘤、自身免疫及代谢性疾病的治疗范式,更驱动了分子生物学、计算生物学与结构表征技术的跨越式演进。鉴于该领域五十载进程之宏大,本文无意提供全覆盖的文献汇编,而是着眼于勾勒行业演进的核心脉络与关键转折。在文献引用上,本文重点关注开创性成果及支撑技术逻辑演进的代表性研究;受限于篇幅,未能尽录其他同行优秀贡献,文中难免有所疏漏,尚祈见谅。
1.1 中心法则与重组蛋白药物研发的科学背景(Scientific Background of the Central Dogma and Recombinant Protein Drug R&D)
二十世纪中叶的一系列关键发现,共同奠定了重组DNA 技术的理论基础。1944 年,美国科学家Oswald Avery,Colin MacLeod 和Maclyn McCarty 的实验证明了DNA 是遗传信息的载体[1]。1953 年4 月,James Watson 和Francis Crick 阐明了DNA 双螺旋结构,两人因此获得了1962 年诺贝尔生理学或医学奖[2]。在此基础上,Francis Crick 于1958 年提出“中心法则”(central dogma),阐明了遗传信息从DNA 到核糖核酸(ribonucleic acid,RNA)再到蛋白质的流动方向[3]。
1961 年,Christian B. Anfinsen 等人通过对核糖核酸酶(ribonuclease,RNase)的变性与复性实验,首次提出了蛋白的一级序列包含了决定其三级结构的全部信息[4]。这一原始发现确立了蛋白质折叠(protein folding)热力学假说的底层逻辑[5],Anfinsen 也因此获得了1972 年诺贝尔化学奖。
1972 年,Paul Berg 等人成功合成了第一个重组DNA 分子,并因此获得了1980 年诺贝尔化学奖[6]。1973 年,Stanley Cohen 与Herbert Boyer 合作,通过质粒载体实现了跨物种的基因重组[7]。基于该成果,全球首家生物技术公司基因泰克(Genentech)于1976 年创立。
真正将效率推向新高度的是Kary Mullis发明的聚合酶链式反应(polymerase chain reaction,PCR) 技术, 该技术从根本上解决了基因获取与复制的效率瓶颈[8]。Mullis 也因此荣获1993 年诺贝尔化学奖。2012 年Jennifer Doudna 与Emmanuelle Charpentier 阐明了CRISPR-Cas9 机制(2020 年诺贝尔化学奖)[9]。2016 年,David Baker 团队在蛋白质从头设计领域取得突破[10] ;2021 年,John Jumper 和Demis Hassabis 领衔开发的AlphaFold 深度学习系统解决了结构预测难题[11],这3 位科学家共同荣获2024 年诺贝尔化学奖。
1.2 重组蛋白药物的基本概念与分类(Basic Concepts and Classification of Recombinant Protein Drugs)
重组蛋白药物是指通过DNA 重组技术,在工程化的宿主系统中生产得到的具有明确治疗功能的蛋白或多肽,具备高特异性、低脱靶毒性等特征[12]。主要分为6 类:①多肽与重组激素类,如人胰岛素、生长激素。②细胞因子类,如干扰素、促红细胞生成素(erythropoietin,EPO)。③重组血液制品、重组酶与凝血因子,如组织型纤溶酶原激活剂[13]、重组人血清白蛋白(recombinant human serum albumin,rHSA)。④单克隆抗体及衍生物,如1984 年诞生的嵌合抗体[14]。⑤重组融合蛋白,如“免疫黏附素(immunoadhesins)”[15]。⑥修饰型重组蛋白,如聚乙二醇化技术[16]。
1.3 重组蛋白里程碑药物的发展史(Development History of Milestone Recombinant Protein Drugs)
(1)重组人胰岛素:1979 年,David Goeddel等人首次在大肠杆菌中表达并折叠出活性人胰岛素[17]。1982 年美国食品药品监督管理局(Food and Drug Administration,FDA) 批准了重组人胰岛素上市,这是全球首个获批的重组蛋白药物,从此胰岛素市场彻底摆脱了对动物胰腺提取的依赖。
(2) 重组人生长激素:1979 年,David Goeddel 等人实现了重组人生长激素高效表达[18]。1985 年,美国FDA 批准了重组人生长激素用于治疗垂体性侏儒症。
(3)重组α 干扰素:20 世纪80 年代初期,随着基因工程技术的突破,科学家成功克隆出人α-2b 干扰素基因[19-20]。1986 年,重组α 干扰素首次获批上市,用于治疗毛细胞白血病。
(4) 重组人EPO :1985 年, 美国安进(Amgen)公司的华裔科学家林福坤及其研究团队首次克隆并在哺乳动物细胞中表达出具有完整生物活性的重组人EPO[21]。该药物于1989 年获得美国FDA 批准。
(5) 贝伐珠单抗:基于Judah Folkman教授1971 年提出的抗肿瘤血管新生(Anti-Angiogenesis) 假说[22],Napoleone Ferrara团队1989 年克隆了血管内皮生长因子(vascular endothelial growth factor,VEGF)[23-24]。抗VEGF 的贝伐珠单抗药物于2004 年获美国FDA 批准用于转移性结直肠癌的一线治疗,并被《科学》(Science)杂志评为2003 年十大科学突破。Napoleone Ferrara 也因此获得了2010 年拉斯克临床医学研究奖。
(6) 重组人血管内皮抑制素:1997 年,Judah Folkman 团队发现具有抗肿瘤活性的内源性蛋白内皮抑制素(Endostatin)[25-26]。由于其特殊的“嵌套式结构”二硫键导致复性极度困难,令全球研发一度停滞。随后,笔者攻克了这一复性难题,并推动改良版重组人血管内皮抑制素于2005 年获批上市。该进展受到国际主流媒体及学术期刊的关注。《华尔街日报》(Wall Street Journal) 于2005 年12 月22 日以头版头条报道了该药物,题为《曾被寄予厚望的抗癌药在中国重获新生》(Once-Touted Drug For Cancer Finds New Life in China), 文中引用了Judah Folkman 教授对该项工作的评价:“ 新版内皮抑制素的问世是罗教授的一大杰出贡献(The new version of endostatin,he says,is an enormous achievement on the part of Prof. Luo.)”[27]。2006 年,《自然·生物技术》(Nature Biotechnology)及《自然·医学》(Nature Medicine)分别对其活性表现和成本优势进行了报道[28-29]。该成果获得了我国2008 年度国家技术发明奖二等奖。目前,重组人血管内皮抑制素仍需每日静脉滴注,提高给药便利性和实现个体化治疗是未来的研发方向[30-31]。作为抗肿瘤血管新生理论的奠基人,Judah Folkman 教授的科学思想在Avastin 和重组人血管内皮抑制素等药物上得到了体现。Folkman 教授于2008 年辞世,其学术贡献对肿瘤治疗领域产生了深远影响。
(7)阿达木单抗:1994 年全人源抗体开发[32]。2002 年阿达木单抗获批上市,用于治疗类风湿关节炎,Greg Winter 荣获2018 年诺贝尔化学奖。
(8)纳武利尤单抗:1992 年Tasuku Honjo团队发现程序性死亡受体1(programmed death-1,PD-1)[33]。2014 年纳武利尤单抗上市,用于治疗晚期黑色素瘤。2018 年,Tasuku Honjo 获得了诺贝尔生理学或医学奖。
(9)德曲妥珠单抗:2016 年第一三共和阿斯利康联合研发了德曲妥珠单抗[34]。2019 年该药物于美国获批上市,用于治疗不可切除或转移性HER2 阳性乳腺癌成人患者。
(10)替泽帕肽:2018 年美国礼来公司的团队研发了葡萄糖依赖性促胰岛素多肽(glucose-dependent insulinotropic polypeptide,GIP)/胰高血糖素样肽-1(glucagon-like peptide-1,GLP-1)激动剂[35]。2022 年该药物于美国获批上市,用于治疗2 型糖尿病。2023 年美国FDA批准用于肥胖或超重成人的长期体重管理。
(11)rHSA :人血清白蛋白是人血浆中含量最丰富的蛋白(一级序列有585 个氨基酸残基,富含17 对二硫键,1 个具有催化能力的自由巯基),约占血浆蛋白总量的60%[36-38],承担了血浆约80% 的胶体渗透压[39-41]。第34 位点半胱氨酸(Cysteine-34,Cys34)残基上的自由巯基约占血液中所有巯基的80%,是血管内最重要的抗氧化基团[42-43]。白蛋白是治疗重大疾病和危急重症的基础药物。但血浆来源的白蛋白存在肝炎、人类免疫缺陷病毒(human immunodeficiency virus,HIV)、冠状病毒等已知或未知病毒感染风险。当前,我国仍面临血浆短缺、“血荒”、注射级白蛋白一剂难求等风险,白蛋白是我国第一紧缺大宗药品,鉴于上述原因,国内白蛋白市场份额60% 以上长期依赖进口[44]。因此,采用生物技术生产重组白蛋白是公认的解决方案。截至目前,我国已有3 家药品企业分别完成了适应症有所差异的Ⅲ期临床试验,所用表达体系包括酵母和水稻。表达体系的不同意味着工艺路线肯定不同,即使同一种表达体系,由于菌种的构建、发酵、纯化甚至复性工艺的不同,也可能会导致产品质量有所差异。因此,不仅要关注重组白蛋白的纯度,还要关注该产品的其他质量属性(如Cys34 残基上的自由巯基保有率、同型半胱氨酸化水平等)与临床长期用药的安全性,如抗药抗体(anti-drug antibodies,ADA)的产生。值得一提的是,笔者团队在国际上率先提出了“年轻、无损、超纯重组白蛋白”的概念,并报道了其研发的“年轻、无损、超纯重组小鼠白蛋白”能够延长小鼠健康寿命[45]、延缓小鼠2 型糖尿病进展[46]。

02
重组蛋白药物常见表达系统( Established Expression Systems for Recombinant Protein Drugs)
重组蛋白药物的成功研发,在很大程度上取决于宿主细胞(表达体系)的选择。理想的表达体系需在生物活性、产量、成本及安全性之间达到精准平衡。
2.1 原核表达系统:以大肠杆菌为主流(Prokaryotic Expression Systems :Escherichia coli as the Mainstream Platform)
大肠杆菌是应用较早、研究较透彻的工业宿主。优点包括:①遗传背景清晰,易于进行基因操纵与载体构建。②生长速度极快,倍增时间短,发酵周期通常以小时计。③产率高且成本低,可实现目的蛋白的高水平表达(常占菌体总蛋白的30%以上),且发酵培养基成分简单、价廉。④包涵体优势,对于某些蛋白,形成包涵体可免受宿主蛋白酶降解,且通过成熟的折叠复性工艺可获得极高纯度的产品。该表达系统的局限:①缺乏翻译后修饰,无法进行糖基化、磷酸化等修饰,不适合生产复杂糖蛋白。②折叠与复性挑战,高表达时易形成无活性的包涵体。通常包涵体蛋白需要在体外经过高效复性后,才能具有成药性。③内毒素污染,作为革兰阴性菌,其细胞壁含有的脂多糖(lipopolysaccharide,LPS)具有强免疫原性,下游纯化去除压力大[47-48]。
2.2 酵母表达系统:以毕赤酵母为代表(Yeast Expression Systems :Pichia pastoris as the Representative Platform)
酵母作为单细胞真核生物,是兼顾生产成本与生物学功能的折中选择。其优点包括:①具备分泌机制,可将目的蛋白直接分泌至培养液中,大幅简化下游纯化工序。②初步的真核修饰,能形成二硫键并进行简单的糖基化修饰。③大规模培养技术相对成熟,易于实现高密度培养,发酵规模可达数万升。该系统也存在如下局限:①重组蛋白易被降解,重组蛋白在分泌过程中和分泌至胞外培养基后皆会受到宿主蛋白酶的降解作用,从而影响重组蛋白的产量和质量。②过度修饰,有时会对蛋白进行过度糖基化(hyperglycosylation),影响蛋白功能[49-51]。
2.3 哺乳动物细胞表达系统(Mammalian Cell Expression Systems)
哺乳动物细胞表达系统主要以中国仓鼠卵巢细胞(Chinese hamsters ovary,CHO) 作为生产复杂蛋白药物的主力。细胞表达体系的优点包括:①修饰精准,具备相对完善的蛋白加工能力,其糖基化修饰模式与人源蛋白高度相似,确保了药物的生物活性和体内药动学(pharmacokinetic,PK)特征。②折叠天然,能在细胞内分子伴侣协助下完成复杂的立体折叠,通常不需体外复性。同时该系统也有以下局限:①生长周期长,倍增时间通常在20h 以上,发酵周期长达10~14 天。②成本高,需要严格的无菌环境、昂贵的培养基和生物反应器设施。③病毒安全风险,存在内源性反转录病毒污染的风险,需进行严格的病毒去除验证[52-55]。
2.4 昆虫细胞- 杆状病毒表达系统(Insect Cell-Baculovirus Expression Vector System,BEVS)
该体系是利用杆状病毒 [ 如苜蓿银纹夜蛾核型多角体病毒(autographa californica nucleopolyhedrovirus,AcMNPV)] 感染昆虫细胞(如Sf9、Sf21 细胞)来生产重组蛋白的技术。常用于疫苗开发及科研用蛋白。该系统优点:①表达量相对较高,可利用强启动子实现外源蛋白的高水平合成。②安全性较好,杆状病毒通常不感染脊椎动物,生物安全风险可控。该系统的缺点包括:①糖基化缺陷,缺乏末端唾液酸修饰,导致蛋白在人体内易被清除。②细胞裂解风险,病毒感染会导致宿主细胞死亡,裂解释放的蛋白酶可能导致目的蛋白降解[56-57]。
2.5 无细胞蛋白合成系统(Cell-Free Protein Synthesis,CFPS)
CFPS 是一种脱离活细胞环境的体外合成技术[58-59]。其主要优点包括:①反应速度快,从基因模板到蛋白产出仅需数小时,适合高通量药物筛选。②开放体系,可添加非天然氨基酸,方便进行定点修饰。但该系统也存在一些局限性:①产量瓶颈,难以实现大规模(如万升级)生产。②能量消耗快, 腺苷三磷酸(adenosine triphosphate,ATP)供应不稳定,副产物积累可能干扰合成过程。③辅助因子缺失:离体环境下难以模拟复杂蛋白质折叠所需的精细微环境[60]。

03
重组蛋白药物常用制备技术(Production Technologies for Recombinant Protein Drugs)
3.1 高密度发酵技术(High-Cell-Density Fermentation,HCDF)
作为提高细胞密度的经典技术,高密度发酵在重组蛋白生产中具有重要应用。鉴于总生产力由细胞密度与比产率共同决定,仅提升单一指标难以实现产量最优化,因此单纯追求细胞密度最大化已非工艺优化的终极目标。Riesenberg 与Guthke指出,HCDF 是在特定时间内于给定体积内最大化产量的先决条件[61-62]。该技术除广泛应用于大肠杆菌和毕赤酵母外[63-64],昆虫细胞和动物细胞(如CHO 细胞)亦有HCDF 的报道[65]。
尽管HCDF 具有显著经济优势,但实施中仍面临底物抑制、氧传递受限、传热障碍、蛋白水解及副产物[ 如二氧化碳(CO2)、醋酸] 积累等局限[62-63,66]。研究多聚焦于建立可放大的发酵体系,解决大规模工艺中纯氧供给、加压反应器、机械负荷及传感限制等问题[67]。其实现主要依靠优化培养条件与控制策略。
HCDF 的优化需统筹培养基设计、环境调控与补料策略3 个方面。培养基设计应遵循限制性原则,精确平衡组分以避免营养过量导致的生长抑制、高浓度沉淀干扰及高离子浓度引发的渗透压应激[67-71]。环境调控方面, 需针对生长期与表达期进行阶段性温度优化以降低信使RNA(messenger RNA,mRNA) 降解[72] ;通过强化传质(如提升通气速率、富氧或加压)克服氧溶解度限制,同时兼顾高氧对蛋白质量的影响[68,73] ;并控制CO2 分压以规避细胞毒性[68,74]。补料策略的核心在于营养供给的动态平衡:以分批补料(fed-batch)作为主流模式,可通过恒速、梯度或指数补料等方式调控比生长速率,其中指数补料能最优维持细胞活性并最大化产物得率[65,67-68,75-85]。灌流技术(perfusion)则借助培养基流动与细胞截留实现极高密度发酵,常用于动物细胞培养。
3.2 分离纯化技术(Separation and Purification Technologies)
在重组蛋白药物的生产流程中,下游纯化(downstream processing,DSP)不仅决定了产品的纯度与安全性,更直接影响其生物活性。以下是生产工艺中常用的分离纯化技术。
3.2.1 离心技术(Centrifugation Technologies)
离心技术是重组蛋白分离的基础技术。20 世纪20 年代,Theodor Svedberg 发明超速离心机并获1926 年诺贝尔化学奖,使蛋白的沉降分析成为可能[86]。在现代工业规模制备中,离心仍是去除细胞碎片和收集包涵体的首选手段[87]。在生物制药工业化进程中,碟片式离心机凭借处理量大、自动化程度高及可线性放大的特性,成为固液分离和澄清工艺从实验室向大规模生产技术转移的关键手段。
3.2.2 色谱技术(Chromatography Technologies)
色谱技术是现代蛋白分离纯化的核心支柱,历经数十年演进形成了各具特色的技术体系。①离子交换色谱(ion exchange chromatography,IEC),通过电荷与固定相的可逆静电吸附实现分离。1956 年Peterson 与Sober 制备的首个工业级离子交换剂标志着该技术正式进入蛋白研究领域[88],其出色的杂质清除能力可针对性去除宿主细胞蛋白、DNA、内毒素、病毒及电荷异构体。②分子排阻色谱(size exclusion chromatography,SEC), 基于分子尺寸差异进行分离。1959 年Porath 与Flodin 发现的交联葡聚糖分子筛效应奠定了其技术基础[89]。因不涉及化学相互作用,SEC 条件温和而能最大程度保持蛋白的天然构象,但受限于处理量与洗脱体积,其在大规模工业制备中的应用正逐渐缩减。目前,多保留于对活性要求极高的精纯步骤。③亲和色谱(affinity chromatography,AC),是目前特异性最高的色谱技术。1968 年 Cuatrecasas等提出的特异性生物相互作用纯化法使其从理论走向实用[90]。例如,Protein A 亲和色谱利用蛋白 A 与抗体 Fc 片段的特异性结合,成为现代抗体工业的首选。既可作为捕获首步,也常用于精制以确保清除微量杂质并保障构象一致性。但其酸洗脱条件可能诱导蛋白聚集,需在下游工艺中严格控制洗脱时间与pH,并随后进行病毒灭活。④疏水作用色谱(hydrophobic interaction chromatography,HIC)与反相色谱(reverse-phase chromatography,RPC)均基于疏水相互作用,但应用场景迥异[91] :HIC 利用蛋白表面疏水区域在特定盐浓度下与配基结合,操作环境高盐温和,对去除蛋白聚集体效果显著。RPC 则通过强疏水作用结合固定相,需要高比例有机溶剂洗脱,具有较高分辨率,在胰岛素商业化生产中常作为精纯步骤将纯度提升至 >99%,但需配备相应的防爆与溶剂回收设施[92]。
3.2.3 切向流过滤技术(Tangential Flow Filtration Technologies)
20 世纪 70 年代后期,超滤膜技术进入生物制药领域,Blatt 等人阐明了其在蛋白浓缩中的应用逻辑[93]。与传统死端过滤(dead-end filtration) 相比, 切向流过滤(tangential flow filtration,TFF)采用料液平行于膜表面流动的操作模式,通过剪切力有效减少浓差极化与膜污染,从而实现连续化、高通量的分离过程。TFF 的核心优势在于其多功能工艺集成能力:既可完成目标蛋白的浓缩与透析,实现缓冲液置换与小分子杂质去除,又能依据分子量截留精度进行粗分离,为后续精纯步骤减轻负荷。在现代生物制药工业化进程中,TFF 实现了大规模生产中的循环高效浓缩与缓冲液置换,现已成为连接各层析步骤的工业“ 交通枢纽” 。

04
蛋白质折叠机制:从遗传密码的后半段到人工智能时代(Protein Folding Mechanisms :From the Second Half of the Genetic Code to the Era of Artificial Intelligence)
分子生物学中心法则描述了遗传信息在 DNA、RNA 与线性多肽链之间的一维流动。然而,生命的功能并非直接取决于这些线性序列,而是始于多肽链折叠成特定的三维空间结构。从一维氨基酸残基序列向三维构象的转化,不仅是表现生命活力所必需的,更被学界深刻地誉为“ 遗传密码的后半段(the second half of the genetic code)”。
如果说决定氨基酸顺序的三联体密码已被破译为“明码”,那么破译一级结构如何决定三级空间结构的规律,则是蛋白质科学中尚未完全揭示的“终极奥秘”。蛋白质折叠描述了多肽链在纳秒至数分钟的时间尺度内,如何从无序伸展态(unfolded state) 精确锁定到唯一天然态(native state)的过程。它涵盖了热力学与动力学、体外与胞内、理论预测与实验验证等多重科学维度。
随着人类基因组计划的完成,蛋白的一级序列数据呈爆炸式增长,而实验测定的空间结构数量却远滞后于序列产出。这种数据上的巨大鸿沟,使得基于 Anfinsen 原理的“ 蛋白结构预测” 成为解决折叠问题的理论核心。理解多肽链如何迅速跨越能量屏障锁定天然态,经历了从宏观假说到微观动力学解析的深刻变革。本章将循着时间脉络,系统回顾这一领域从实验奠基到人工智能(artificial intelligence,AI)预测的里程碑进展。
4.1 奠基时代:Anfinsen 热力学假说与Levinthal 构象悖论(The Foundational Era :Anfinsen's Thermodynamic Hypothesis and Levinthal's Paradox)
20 世纪50 至60 年代,蛋白质折叠研究正式开启。1961 年,Christian B. Anfinsen 通过牛胰腺RNase A 的经典变性复性实验证明,蛋白的一级序列包含了决定其三级结构的全部信息[4]。他在1972 年获诺贝尔化学奖时的演讲中系统提出了“热力学假说(thermodynamic hypothesis)”, 强调天然态处于自由能最低点[5]。这一假说不仅揭示了生命信息的表达逻辑,更是所有蛋白复性技术的理论前提。
然而,折叠的实现不仅是热力学驱动,更受动力学制约。Cyrus Levinthal 提出了著名的Levinthal's Paradox :若多肽链仅靠随机尝试所有构象来寻找正确结构,其耗时将超越宇宙寿命。这在逻辑上推导出折叠必经特定的有序“路径(pathways)” 而非盲目碰撞[94-95]。由于Levinthal 构象悖论的原始文献未被PubMed 收录,后世多通过Zwanzig 等人的评述深入理解这一悖论[96]。至此,热力学与动力学的双重逻辑共同构筑了蛋白质折叠研究的基石。在笔者看来,这一时期的贡献不仅在于给出了答案,更在于定义了此后半个世纪我们所追逐的“ 终极奥秘”:序列是如何在有限时间内锁定唯一结构的?
4.2 动力学探索时代:折叠中间体理论的建立与演进( The Kinetic Exploration Era: Establishment and Evolution of Protein Folding Intermediate Theory)
为破解经典的Levinthal 构象悖论,学术界开始探究蛋白质折叠路径中是否存在关键的中间体(intermediate)。尽管在这一时期,蛋白质折叠机制研究先后孕育出多种各具特色的理论假说,但从指导重组蛋白复性实践的维度审视,中间体理论无疑是最具核心的工具价值。受限于篇幅,本部分不拟对各类模型进行冗余评述,而将聚焦于中间体理论的演进历程。本部分将结合作者在该领域的基础理论研究,以及后续在重组蛋白药物研发、工业化大规模复性技术开发中的科研实践,深度探讨其内在的逻辑本质,即如何从一级氨基酸残基序列信息中,解码蛋白的三级空间折叠逻辑。这一过程,实质上构成了生命科学中“遗传密码的后半段”的核心内涵。
4.2.1 折叠中间体的实验发现(Experimental Discovery of Folding Intermediates)
20 世纪70 年代初,蛋白质折叠研究迎来了重大的理论转移,研究重心开始从经典的“两态模型(two-state model)”向“多步动力学路径(multi-step kinetic pathway)”理论转型。
蛋白质动力学路径的研究,首先建立在对变性状态物理性质的精确定义之上。C. Tanford 等通过对多种蛋白在强变性剂中理化性质的系统表征,证实完全变性后的蛋白呈“无规卷曲(random coil)”状态[97]。这一结论界定了折叠起始点的物理边界,为探寻非随机折叠路径提供了必要的基准坐标。
在此背景下,R. L. Baldwin 成为动力学中间体研究的先驱。1971 年,他通过观测RNase A的连续变性动力学,率先捕捉到了折叠过程中快速初始阶段的存在[98]。随后在1972 年,他论证了中间态存在的顺序模型(sequential model)[99],打破了单一的一步折叠观。1973 年,Baldwin 团队进一步证明,RNase A 无论经由快反应还是慢反应路径,最终均能产生具有生物活性的天然酶[100],为“折叠路径”的多样性提供了早期的实验依据。
1974 年,T. E. Creighton 另辟蹊径, 以还原态牛胰蛋白酶抑制剂(bovine pancreatic trypsin inhibitor,BPTI) 为模型, 利用化学淬灭法成功捕捉并分离出瞬态共价折叠中间体[101],证实折叠遵循由特定二硫键依次形成的动力学路径[102],且部分包含非天然二硫键的中间体需经异构重排方能完成最终折叠[103]。值得注意的是,Baldwin 与Creighton 在同一时期分别通过非共价动力学观测与共价键捕捉这两种截然不同的实验体系,共同指向了中间体的存在。这种“ 殊途同归”的研究结论,促使学术界对折叠机制的认识从单一的“两态模型”转变为多步的“路径”假说。
这一阶段的研究成果最终汇聚于Baldwin 在1975 年发表的里程碑式综述中,系统确立了中间体在蛋白质折叠机制中的核心地位[104]。Baldwin的学术整合能力打破了早期的“全或无(all-or-none)”模型。他指出,动力学研究显示的“ 多相性”意味着反应过程中至少涉及3 种以上的状态。通过对BPTI 蛋白中二硫键中间体的深度分析,他阐明了研究者可以直接观察折叠路径中二硫键对构象形成的驱动作用。随着快速反应技术的应用,学术界开始捕捉到更为瞬时和微妙的中间体信号。Baldwin 指出即使在看似完全展开的状态中,也可能保留了特定的局部结构,所有这些研究的终极目标是探寻折叠的立体化学决定因素。同年,针对折叠中的慢反应步骤,J. F. Brandts 等提出这可能源于肽链中脯氨酸残基的顺反异构化[105],该观点在Baldwin 1975 年发表的综述中得到了重视:不仅解释了Baldwin 观测到的慢反应现象,也为后来发现肽基脯氨酰顺反异构酶(peptidyl-prolyl cis-trans isomerase)奠定了理论基础。
4.2.2 折叠中间体理论的建立(Formation of the Folding Intermediate Theory)
关于蛋白质折叠中间体的权威性论述,尤其是动力学中间体的系统理论,主要源于R. L.Baldwin 及其学生Peter S. Kim 撰写的两篇里程碑式综述。
(1)从假说到模型:1982 年的理想主义探索(From Hypothesis to Model :Idealistic Explorations in 1982)。 在1982 年发表的综述中[106],Baldwin 和Kim 系统探讨了小分子蛋白质折叠的机制:明确提出折叠并非简单的两态过程,并给出了经典的U(变性态)→ I(中间体)→ N(天然态)三态模型;提出“框架模型(framework model)”,认为二级结构的形成领先于三级结构的组装,极大地缩减了构象搜索空间;并提出多结构域蛋白的各结构域通常独立折叠。Baldwin 提出“折叠路径是破解序列决定结构之谜的关键密码”,这一理想化的提法吸引了包括本文作者在内的无数科学家投身于此。
(2) 从模型到现实:1990 年的理论成熟与动态组装路径(From Model to Reality :Theoretical Refinement and Dynamic Assembly Routes in 1990)。时隔8 年,Kim和Baldwin 于1990 年再次发表综述[107],标志着中间体理论正式进入成熟期。文章深度解析了“熔球体(molten globules)”这一关键中间态(该术语最初由Ohgushi 和Wada 命名[108], 并由Ptitsyn 系统解释[109])。Kunihiro Kuwajima 进一步阐释了“熔球体”作为理解结构协同性的关键线索,代表了变性多肽发生疏水崩塌后的一个关键能量台阶[110]。这种物理学洞见在Kuwajima 晚年提出的“两阶段层级模型”中得到升华[111]。Yuji Goto 等人通过电荷屏蔽理论证明阴离子可稳定酸诱导的熔球态(molten globule,MG)[112],这为工业化蛋白复性工艺中预防蛋白聚集提供了极具价值的指导。C. Robert Matthews 对不同家族蛋白质折叠路径的一致性进行了系统综述[113]。S. Walter Englander 团队则利用氢氘交换技术捕捉瞬时中间体[114],提出“顺序稳定机制”和“折叠子(foldons)”理论[115],并在2023 年将其升华至对复杂分子机器运转的全局解释[116]。至此,成熟期的理论确立了蛋白的最终结构由热力学稳定性决定。
4.3 能量景观与“折叠漏斗”:从“单一路径”到“统计热力学”的转移(Energy Landscape and Folding Funnel: The Shift from "Single Pathway" to "Statistical Thermodynamics")
承接对折叠中间体和特定路径的深入探讨,学术界逐渐认识到,蛋白质折叠并非总是严格遵循一条狭窄的“单行道”。20 世纪90 年代,以PeterWolynes、Ken Dill 和José Onuchic 等人为代表的科学家引入了统计物理学概念,提出了“能量景观理论(energy landscape theory)”及具有启发性的“折叠漏斗(folding funnel)”模型[117]。这一理论标志着蛋白质折叠研究从探寻“单一微观动力学路径”向“宏观统计热力学”的重大跨越。
漏斗的三维形貌与热力学驱动力:在漏斗模型中,纵坐标代表大分子的内部自由能,横向宽度代表构象熵。随着疏水崩塌发生,多肽链向内部紧缩,构象熵急剧减少,自由能不断下降,犹如水流顺着漏斗向谷底汇聚,最底部即为唯一热力学稳态——天然构象。
终结Levinthal 构象悖论的理论利器——漏斗模型指出:蛋白绝非进行盲目的全局随机搜索,而是受到能量地形的偏倚引导[118]。多条平行的微观组装路径如同百川入海,迅速向着能量最低点收敛。
漏斗的“粗糙度”与体外复性的物理学解释:真实的折叠漏斗内壁充满了能量起伏,形成大量局部极小值,这正是MG 在能量景观上的投影[119]。在体外高浓度环境下,如果局部能量陷阱过深,多肽链就会陷入其中,形成动力学陷阱(kinetic traps),导致错误折叠甚至不可逆的聚集。这从根本上解释了为何重组蛋白体外复性往往面临极大的收率挑战。
连接未来的理论基石:能量景观不仅统一了争论,也为分子伴侣机制提供了理论基础。科学家开始尝试用计算手段解析能量地貌[120],这口“漏斗”蕴含的物理法则,正是几十年后AI 深度学习隐式重构的核心。
4.4 熔球态的协同性争议与二硫键的“双刃剑效应”(Cooperativity Debate of the Molten Globule State and the "Double-Edged Sword" Effect of Disulfide Bonds)
4.4.1 学术分歧的缘起:关于熔球态协同性的认知冲突(Origins of Academic Divergence: Cognitive Conflicts Regarding Molten Globule Cooperativity)
在蛋白质折叠研究领域的发展历程中,MG中间体在变性与复性过程中是否具备协同性(即二级结构与三级结构同步变化),曾引发一场影响深远的学术争论。20 世纪90 年代中期,Kim 团队及其合作者连续报道,α- 乳清蛋白(alpha-lactalbumin,α-LA)的MG 不具有协同性[121-122];而笔者与Baldwin 则通过研究证实,脱辅基肌红蛋白(apomyoglobin,apoMb)中间体呈现出高度协同性[123]。这种实验结论的对立,曾使研究陷入深度逻辑困境。作为Baldwin 学术团队的核心成员,笔者通过设计严谨的实验方案,不仅化解了危机,更揭示了二硫键对折叠动力学的深层调控机制。为此,我们提出核心假设:不一致究竟源于检测方法差异,还是模型蛋白本身的结构差异?
4.4.2 检测方法的验证与排除(Validation and Exclusion of Detection Methods)
Kim 团队采用的是高分辨二维核磁共振(high-resolution two-dimensional nuclear magnetic resonance,2D NMR),而笔者采用了更为简洁高效的“叠加测试”(superposition test)——通过圆二色光谱(circular dichroism,CD)测量α- 螺旋二级结构,与色氨酸内源荧光发射光谱测量三级结构,同步监测。笔者首先利用该方法对Kim 团队所用的α-LA 突变体进行验证,确认其确实呈现非协同性特征,从而排除了检测方法差异的影响。
4.4.3 二硫键的“双刃剑效应”(The "Double-Edged Sword" Effect of Disulfide Bonds)
将焦点锁定于模型蛋白结构差异,Kim 团队所用α-LA 突变体含有一对天然二硫键(Cys28-Cys111)。本文作者预判:正是这对二硫键“锁定”了局部构象,导致MG 呈现非协同性。当通过定点突变去除该二硫键后,α-LA 中间体立即展现出典型的协同折叠模式[124]。Kim 团队随后报道在无二硫键约束下,α-LA MG 同样具有协同性[125]。
更为关键的实证来自于一组跨越时光的原始实验数据:借鉴前人确定的脱辅基肌红蛋白(apoMb)核心结构[126],笔者在原本呈现高度协同性折叠的apoMb 核心区域人为引入两个 Cys 以构建人工二硫键。实验结果(图1)直观显示,原本高度协同的野生型(WT)apoMb 中间体[123] 在引入一对二硫键后(apoMb 突变体H24C-H119C)立即转变为非协同折叠模式,因为代表二级结构的CD与代表三级结构的色氨酸荧光发射光谱(FL)变化曲线非重合(非同步性或非协调性);而且野生型的折叠曲线非常陡峭,呈现高度协同性;而含有二硫键突变体的折叠曲线则变得极为平缓宽泛。这直观地证明了人为引入二硫键后,物理约束确实显著削弱了折叠的协同性。基于此双向验证,笔者提炼出一条普适性物理化学规律:不含二硫键的折叠中间体通常具有高度的协同性;而二硫键虽能显著稳定蛋白质的天然构象,但由于其引入的物理约束,反而会削弱中间体的折叠协同性。此即为二硫键在蛋白质折叠中所展现的“双刃剑效应”。
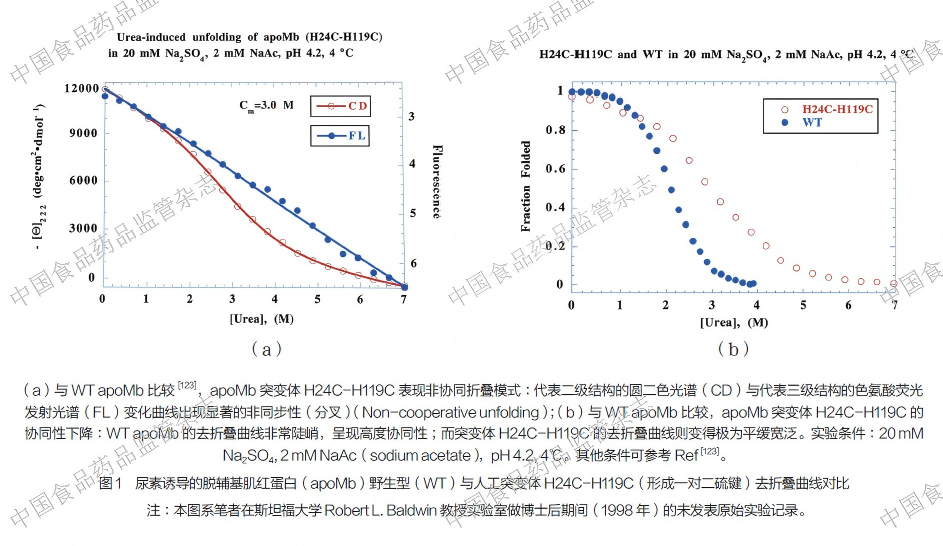
4.4.4 理论向应用的延伸:重组蛋白药物的工业化复性(Extension from Theory to Application: Industrial Refolding of Recombinant Protein Drugs)
这一效应在重组蛋白药物研发中意义重大:①作为“结构锁扣”,显著提高天然态的热力学稳定性,延长药物有效期。②作为“动力学枷锁”,在体外复性中,二硫键极易诱导肽链偏离协同折叠路径。从前述能量景观视角看,这相当于使多肽链跌入难以逾越的动力学陷阱,导致误折叠和聚集。
这一发现为笔者团队日后攻克数种含有复杂二硫键药物及含有自由巯基的肿瘤标志物的工业化复性难题提供了核心理论指导。通过人为规避“枷锁”效应,引导高效协同折叠,最终实现了复杂药物的成功产业化。
4.5 细胞内的蛋白质折叠“机器”:分子伴侣与折叠酶的护航(Intracellular Protein Folding "Machinery": Chaperones and Foldases)
在真实的、高度拥挤的胞内环境下,刚刚合成的多肽链极易发生非特异性相互作用。因此,体内折叠受到一套精密“分子机器”的辅助。①分子伴侣。跨越聚集陷阱的“隔离容器”, Hartl 揭示了分子伴侣(GroEL/ GroES 复合体)辅助折叠的精妙机制[127]。它们形成纳米级“容器”,为部分折叠多肽提供隔离微环境[128],有效防止多分子交叉聚集,协助蛋白跨越能量陷阱。②折叠酶。突破能量瓶颈的“生化加速器”,内质网中配备了“折叠酶”加速缓慢步骤。对于蛋白质二硫键异构酶(protein disulfide isomerase,PDI),1963年Venetianer 和 Anfinsen 团队分别独立发现其能重新激活还原态酶[129-130] ;1985 年Edman等人首次克隆其互补DNA(complementary DNA,cDNA) 序列[131]。对于PPIase,1984年Fischer 首次证实其活性[132] ;此后亲环蛋白被克隆并最终被证实与PPIase 为同一蛋白[133-135]。异构化过程的加速极大提升了整体折叠效率。
4.6 错误折叠与疾病:从朊病毒到淀粉样变性(Misfolding and Disease :From Prions to Amyloidosis)
多肽链错误陷入折叠漏斗深度陷阱不仅会丧失功能,更会引发病理反应。1997 年诺贝尔生理学或医学奖得主 Prusiner 提出的“朊病毒”首次证明错误构象可致病且具传染性[136]。随后Dobson 系统阐述了淀粉样变性疾病的通用机制,指出形成高度有序、富含β- 折叠的淀粉样纤维是能量景观中的另一种极低能量聚集态[137]。这揭示了生理功能与病理毒性间微妙的热力学博弈。
4.7 硅基智能的巅峰:AlphaFold 与AI时代(The Pinnacle of Silicon-Based Intelligence :AlphaFold and the AI Era)
从Anfinsen 提出假设,到折叠中间体理论的建立,再到确立能量景观与分子伴侣网络,这段探索历程迎来了硅基智能的破局。2024 年诺贝尔化学奖宣告了AI 时代的到来。以AlphaFold 为代表的深度模型,以前所未有的精度隐式重构了前人积累的折叠法则[137-138],实现了高精预测[139],并为从头设计(de novo design)打开可能——自Baker 团队设计Top7 蛋白[10],到基于扩散模型的定制化生成[140]。更进一步,AlphaFold 3 打破了单体预测壁垒,能够高精度预测大分子互作网络[141],标志着硅基智能正式迈向全景式模拟微环境的新纪元。
4.8 蛋白质折叠机制探索结语与展望:在硅基预测之外的碳基重塑(Summary and Perspectives on Protein Folding Mechanisms: Carbon-Based Remodeling Beyond Silicon Prediction)
从Anfinsen 那个简洁而深邃的“热力学假说”诞生至今,人类探索蛋白质折叠机制的征程已逾一甲子。回顾这段波澜壮阔的历史,不难发现,每一步理论的跨越都伴随着实验技术的迭代更新:从NMR 对中间体的捕捉,到统计力学对能量景观的重构;从分子伴侣对细胞质量控制网络的揭示,再到如今AlphaFold 等深度学习算法对蛋白三维构象的精准预测。蛋白质折叠不再仅仅是一个关于氨基酸残基如何排列的物理化学课题,它已演变为连接遗传信息(一级结构)与生命功能(三级空间结构)的“生命第二密码”。
然而,AI 对蛋白结构预测范式的彻底颠覆,并不意味着蛋白质折叠这一“世纪难题”的终结,而是一个全新维度的开启。正如早期折叠动力学开拓者们所坚守的那样,“高精度的构象预测”并不完全等同于“底层物理机理的阐明”。硅基智能能够以前所未有的速度解析海量结构,但结构预测的终点恰恰是物理实体干预与解析的起点,尤其是对于结构高度不均一且处于动态瞬变过程中的蛋白折叠中间体而言,实证物理研究依然不可替代。
一方面,尽管硅基智能已能以前所未有的通量隐式利用序列中的协同性规律,但在解析多肽链如何跨越特定能垒、如何通过瞬时中间体寻找到天然态的动态路径(folding pathway)上,“碳基科学家”仍保留着不可替代的解释权。这不仅是目前算法的局限,更是生命科学从“静态快照”向“动态演化”跨越的必经之路。
另一方面,更深层的折叠机制对生物医药工业的持续赋能,具有极其重要的现实意义,这也是重组蛋白药物研发半个世纪以来的核心诉求。重组蛋白药物的工业化制备曾长期受限于动力学层面的复性瓶颈。随着对疏水崩塌、动力学陷阱机制理解的深入,生物工艺正逐步告别传统的“试错法(trial and error)”,转向由底层物理规律驱动的精准调控。当我们能够跨越庞大分子量与复杂二硫键的折叠鸿沟,研发出“年轻、无损、超纯”重组白蛋白(PG-rHSA)这般纯净、几乎未受任何非酶促修饰(如Cys34 的自由巯基、糖化、羰化、同型半胱氨酸化)的“纯天然构象”时,面对单瓶高达10 g 大剂量的临床需求,我们不仅是在制造一种高质量的生物药,更是在系统性地重塑生命的微环境。这种基于底层折叠机制的结构重置,为逆转机体衰老、修复微循环损伤,提供了AI 无法在虚拟世界中直接生成的“真实解药”。
半个多世纪以来,几代“蛋白质折叠者”致力于破译这门从微生物、植物、动物到人类共享的底层科学语言 [DNA,RNA,多肽与蛋白质折叠(polypeptide and protein folding)] 。站在AI时代的历史节点回望,科学的终极目标并非仅仅是在计算机中获得一个完美的预测模型,而是用完美的天然构象去对抗岁月的熵增。破译并应用这门语言的三维奥秘,不仅是为了治愈疾病,更是为了彰显整个碳基世界生生不息的尊严。这正是人们不懈探索蛋白质折叠机制的终极意义,也是那份近乎执拗的坚守背后,最深沉的回音。

05
重组蛋白药物常见复性工艺与试剂(Established Refolding Processes and Reagents for Recombinant Protein Drugs)
5.1 主流复性技术(Mainstream Refolding Technologies)
无论采用何种表达系统,重组蛋白药物研发的最大难点之一在于实现正确复性,即形成正确的空间结构并具备生物学功能。将水溶性的重组蛋白想当然地等同于正确复性,是一个长期存在的误区,且这一问题长期以来未引起足够重视。
蛋白质复性是以结构域(structural domain)为单位的。通常分子量越大、二硫键数目越多,复性越困难;其中最具挑战性的是富含二硫键且存在游离巯基的蛋白质。掌握复性核心工艺是提升产品竞争力的关键,尤其对于包涵体来源的蛋白质。包涵体通常是外源基因在原核宿主中高水平表达时,因折叠速率滞后于合成速率、缺乏辅助因子(如分子伴侣)或氧化还原环境不适,而在细胞内形成的不可溶性蛋白聚集体。其优势在于高表达量及便于回收,在工业规模生产中具有显著吸引力。然而,核心瓶颈在于将变性蛋白质重折叠(复性)为具有生物学活性的天然构象——该步骤仍是制约产业化的关键技术壁垒。
包涵体遵循分离纯化、变性溶解、粗纯化、复性、复性后精纯化策略:先分离纯化包涵体,再将纯化后的包涵体(目标蛋白质纯度约为90%)使用高浓度的变性试剂 如尿素、盐酸胍、十二烷基硫酸钠(sodium dodecyl sulfate,SDS)或十六烷基三甲基溴化铵(Cetyl trimethyl ammonium bromide,CTAB) 等破坏聚集体中的相互作用,并加入还原剂 如二硫苏糖醇 ( dithiothreitol,DTT)、三(2- 羧乙基)膦(Tris(2-carboxyethyl)phosphine,TCEP)等打开错误形成的二硫键,使目标蛋白完全伸展为变性状态;然后对变性状态的目标蛋白进行纯化,去除宿主细胞蛋白(host cell proteins,HCP)、核酸、内毒素等干扰因素;之后逐步降低或去除变性试剂和还原剂,促使目标蛋白重新折叠并恢复其天然构象,完成复性(该步骤本身也具备内在纯化作用);最后对复性后的目标蛋白再次精细纯化,获得具有正确空间构象和完整生物学功能的高纯度蛋白。需要指出的是,即使目标蛋白自身不含二硫键或自由巯基,在变性溶解包涵体时仍应加入还原剂,以防止包涵体中残留的HCP 因自身巯基氧化而形成非特异性交联或聚集。
复性是整个策略的核心步骤,其方法主要包括以下几类。
(1)稀释复性:将高浓度变性溶液直接或缓慢滴加到大量复性缓冲液中,瞬间或快速降低变性剂浓度,促使蛋白重新折叠。该方法操作简单,但复性率低且体积急剧增大,导致后续浓缩和纯化困难,在工业化生产中应用受限。
(2)透析复性:利用半透膜将小分子变性剂逐步扩散出去。该方法条件温和,适合于剪切力敏感的蛋白。现代工艺多采用压力或离心驱动下的超滤膜连续置换技术,可在浓缩的同时进行复性,提高了效率并控制了体积膨胀,但对设备和膜材质有较高要求。
(3)色谱复性:通过色谱介质与变性蛋白相互结合,在吸附状态下去除变性试剂促使蛋白质折叠。常见的色谱方法包括IEC、HIC 及SEC 等。该方法将复性和纯化整合,利用空间隔离效应解决蛋白聚集的问题,但操作相对复杂而且介质成本较高。
高压条件可以通过物理方式使蛋白变性,同时抑制聚集,提高复性收率[142-144]。沸石作为一类结构均一的多孔硅酸铝晶体,能在6 mol/L 盐酸胍溶液存在下吸附蛋白,随后在复性缓冲液实现洗脱和复性[145-146]。科研人员仍在持续开发新型蛋白复性方法以建立简洁且经济高效的工艺。
尽管复性的核心目标是获得蛋白的天然构象,但许多功能蛋白的分子构象本身具有动态可塑性。黄成栋团队利用液体NMR 技术证实,分子伴侣热休克蛋白90(heat shock protein 90,Hsp90)具有多种构象动态转变特征,并阐明了其功能实现依赖于不同构象状态之间的转换[147]。Hsp90α 传统上被视为肿瘤治疗的胞内靶点,后续研究发现其可被分泌到胞外发挥功能[148-150],并在血浆中成为一种泛癌标志物[151-152]。上述发现使得靶向分泌性Hsp90α 的药物开发策略提供了广阔的应用前景。其中,单克隆抗体因其高特异性和优良的PK 特性受到广泛关注,多个研究团队已证实靶向分泌性Hsp90α 的单克隆抗体能有效抑制肿瘤生长和转移[150,153-156]。但在抗体药物开发过程中,除了关注Hsp90α 纯度外,必须高度重视其构象均一性,通过构象锁定策略或表位定向筛选,确保抗体对功能相关构象的识别效率与治疗特异性。
除大肠杆菌外,酵母、杆状病毒– 昆虫细胞及哺乳动物细胞等表达系统同样存在蛋白质错误折叠与聚集问题。可以通过分子伴侣与折叠酶共表达、基因工程调控分泌通路及培养工艺优化等策略提升胞内折叠效率。但是无论采用何种表达平台,一旦目标蛋白无法在胞内完成正确折叠,最终仍需依赖体外复性获得功能性构象。不同表达系统间的体外复性原理与核心策略具有较高共性,体外复性已成为重组蛋白药物开发中的关键共性问题,也是本部分重点讨论的核心内容。
5.2 常见复性添加剂与辅助试剂(Common Refolding Additives and Promoting Agents)
在变性蛋白(U)的复性过程中,通常先形成折叠中间体(I),再进一步折叠为天然态(N)[157-159]。由于中间体(I)容易使折叠路径偏离并引发聚集,正确的折叠路径与错误的聚集路径之间存在动力学竞争。因此,复性缓冲液中需引入特定试剂以提高活性蛋白得率[160-161]。
(1)氧化还原类试剂:对于含二硫键蛋白,构建合适的氧化还原环境 ,如氧化型谷胱甘肽(glutathione disulfide,GSSG)/ 还原型谷胱甘肽(glutathione,GSH)、DTT/GSSG 至关重要[162]。通常控制体系呈弱还原性,并在弱碱性环境下进行,有利于二硫键的形成与重排[160-163]。
(2) 有机共溶剂:二甲基亚砜(dimethyl sulfoxide,DMSO)是一种弱氧化剂,可在 pH3~8 范围内促进多肽二硫键的形成[164]。该类试剂兼具优良的化学选择性和水溶性:既不会引发其他亲核性氨基酸的氧化副反应,又可通过提高浓度显著加速二硫键配对反应,因而被视为高效的复性添加剂。此外,DMSO 能够削弱疏水效应,减少因疏水斑块过快或过强结合所导致的蛋白聚集[165]。DMSO 的折叠辅助效应具有典型的双相浓度特征,即在较低浓度下(通常低于10%~20%)表现出稳定或促进折叠的作用;但是当其浓度超过阈值时,表现为强变性剂效应。
(3)低浓度变性剂:低浓度的尿素和盐酸胍能有效抑制中间体(I)的聚集,有利于复性[158,166]。
(4)氨基酸类:精氨酸可与蛋白之间发生弱的相互作用,抑制蛋白聚集[167-168] ;精氨酰胺[169]、甘氨酰胺[170] 均被报道能显著提高复性效率。脯氨酸可与中间体(I)结合,成为不易聚集的结构单元,保护蛋白正确折叠[171]。
(5)表面活性剂:如3-[3-(胆酰胺丙基)二甲氨基] 丙磺酸内盐(3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate,CHAPS)、曲拉通X-100(Triton X-100)、CTAB、聚乙二醇(polyethylene glycol,PEG)等可遮蔽中间体(I)暴露的疏水表面,防止聚合[172-174]。但要严格控制其浓度以避免干扰蛋白质折叠,或加剧聚集现象[175]。此外,纯化后残留的表面活性剂可能影响后续的结构表征或功能测定,因此在实验设计中需预先评估去除策略。
(6)环糊精及其衍生物:其疏水空腔可与中间体(I)的疏水区域形成非共价包合物,阻断分子间疏水作用来抑制聚集[176-177]。人工伴侣系统[178]巧妙地利用环糊精的疏水空腔,抽提变性蛋白上的表面活性剂,促使蛋白在受控环境中启动自主折叠,模拟天然分子伴侣的作用。
(7)其他类试剂:甘油、海藻糖及PEG 可降低分子碰撞概率,稳定蛋白的结构[173,179-180]。
工业化级复性必须同时满足“四高”基准才具备产业化价值:高复性浓度(>1 g/L,此时变性的目标蛋白纯度不应该低于95%)、高复性率(>50%)、高复性收率(>50%)及复性后的再纯化使纯度达到要求(>99%)。对于临床剂量大的重组蛋白药物,为实现药品价格的可及性,复性浓度需远高于1 g/L,同时复性率需超过50%,甚至达到90% 以上。需要注意的是,复性率和复性收率是两个不同的概念。内皮抑素(Endostatin)的产业化突破即为典型案例。该蛋白是一种分子量约20 kDa 的内源性血管生成抑制剂,其结构主要由不规则环和β- 折叠片构成,仅含少量α- 螺旋,尤为关键的是包含了两对嵌套二硫键(nested disulfide bonds)[181-182]。这两对二硫键的正确配对是维系Endostatin 空间结构及抗血管生成活性的核心,也正是该蛋白复性极具挑战性的结构根源。发现Endostatin 的哈佛大学Folkman 实验室在早期研究中,复性体系里该蛋白浓度仅为0.1 mg/ml,复性效率 <1%[25],制备成本高昂且产物稳定性差,完全不具备工业化可行性。基于对蛋白质折叠机制的深刻理解,笔者于1999 年12月25 日成功解决了这个复性难题[183]:实现了E.coli表达的Endostatin 包涵体蛋白复性浓度 >1 g/L、复性率 >50%、复性后的Endostatin 再纯化的纯度 >99% 的工艺指标,解决了二硫键正确配对问题。该产品于2005 年获准在中国上市,成为包涵体来源重组蛋白药物成功产业化的里程碑事件。需要说明的是,该复性工艺的具体配方及参数尚未在学术文献中公开,仍属商业机密。同年笔者实验室进一步揭示了Endostatin 的两对嵌套二硫键的独特功能分工:Cys135–Cys165 是维持三级结构完整性的核心,而Cys33–Cys173 则像一把锁,限制了多肽骨架过度形成的α- 螺旋倾向[184]。这种二硫键嵌套模式也阐明了该蛋白极强的耐酸性的结构基础[185]。
5.3 复性产物的质量评价与验证(Quality Evaluation and Validation of Refolded Products)
判断复性成功与否仅观察可溶性是远不够的,水溶并不意味着成功复性,但成功复性后的蛋白一定是水溶的(个别膜蛋白可能除外),必须通过一套多尺度的分析与表征技术来验证[183-187]。
(1)电泳技术:SDS- 聚丙烯酰胺凝胶电泳(SDS-polyacrylamide gel electrophoresis,SDS-PAGE)通过比较还原性与非还原性条件下的电泳迁移率差异,可判定复性后蛋白分子内/ 分子间二硫键的形成状态及连接模式。在常规质量控制中,考马斯亮蓝染色法应用广泛。通常要求在还原性电泳条件下,每泳道上样量为5 μg 时,获得清晰的单一条带,该指标较易实现。但在非还原条件下,实现相同上样量获得清晰单一条带的指标有一定的难度。若非还原条件下出现微弱杂带,建议采用灵敏度更高的银染法(灵敏度≤ 1 ng)进行确证。Switzer 等在1979 年首次在聚丙烯酰胺凝胶体系应用了银染法,实现了比考马斯亮蓝染色高约100 倍的灵敏度,并奠定了现代蛋白银染技术的基础[188],当前银染法依然是聚丙烯酰胺凝胶中蛋白检测最灵敏、最可靠的方法之一[189]。我们的经验是,当分别在还原性与非还原性条件下,每泳道上样1 μg 实现银染单一条带时,表明复性后的蛋白纯度可满足绝大多数重组蛋白药物后续结构表征与功能研究的基本要求。
(2) 高效液相色谱(high performance liquid chromatography,HPLC):高效分子排阻色谱(SEC-HPLC)是评估蛋白复性状态的有效方法之一。成功复性的蛋白应在SEC 图谱上呈现单一、对称的洗脱峰。若出现多峰现象,应优先关注最先出现的尖锐、对称的特征峰,其余前、后伴行的“ 馒头峰” 或肩峰等多是寡聚体、异构体或折叠中间体。蛋白在还原性和非还原性条件下的色谱迁移速度存在差异,还原状态下蛋白结构松散、流体力学体积增大,迁移速度变慢,利用这一差异可有效判断二硫键的形成状态。高效反相色谱(reverse-phase HPLC,RP-HPLC) 可检测因错误折叠导致疏水内核暴露的构象变体。正确折叠的蛋白中,疏水氨基酸残基通常包埋于分子内部;而错误折叠或部分去折叠时,暴露的疏水基团与色谱柱的固定相(如C18)的结合能力更强,需更高浓度有机溶剂才能洗脱,表现为保留时间延长、出峰更晚。高效离子交换色谱(ion exchange HPLC,IEX-HPLC)则用于监测电荷异质性,其原理基于带电荷的蛋白与带相反电荷的色谱柱固定相之间可逆静电相互作用,利用表面净电荷差异在特定pH 下分离并区分出不同电荷变异体。
(3) 荧光发射光谱:自1958 年Szent-Györgyi 首次确立色氨酸作为蛋白内源荧光探针以来,色氨酸荧光已从单一的光谱观测演变为能够解析蛋白局部结构、动力学、相互作用及药物效应的综合工具[190-191]。色氨酸作为疏水性氨基酸,通常位于蛋白内部,在288 nm 处具有吸收并能被激发产生荧光。因此,在288 nm 激发下,记录蛋白在300~400 nm 范围内发射的荧光光谱,可有效判定其折叠状态:当色氨酸残基从水相环境埋入蛋白非极性核心时,其最大发射波长发生蓝移,指示三级结构趋于致密、天然构象形成。需指出的是,色氨酸并非始终位于蛋白内部,部分可暴露于分子表面,但是这一局限可借助AlphaFold 等结构预测工具预先评估其空间位置[10,137-141],以辅助数据解读。
外源8-苯胺基-1-萘磺酸(8-anilino-1-naphthalenesulfonic acid,ANS)作为蛋白疏水位点的荧光探针,最早由Weber 于1964 年系统建立[192],此后该探针逐渐发展为检测蛋白三级结构的折叠致密性与局部疏水区域的暴露程度的常用工具[193-194]。ANS 探针特异性结合于蛋白表面暴露的疏水斑块,其荧光强度增强表明蛋白结构松散、疏水核心未充分包埋。综合色氨酸内源荧光与ANS 外源探针的信号变化,可提供更全面的折叠动力学及构象稳定性信息。
(4)CD :直接定量蛋白的二级结构组成,精确追踪α- 螺旋、β- 折叠等含量的变化。
(5)NMR :验证蛋白结构的“金标准”,揭示蛋白的瞬态结构与动力学特征。
(6)等电聚焦:测定蛋白质的等电点(isoelectric point,pI),可灵敏地检测复性过程中因构象变化或化学修饰引起的电荷异质性,验证蛋白表面电荷分布的正确性,确保复性产物的构象真实性与批次一致性。
(7)质谱(mass spectrometry,MS)技术:精确鉴定分子量、序列、二硫键配对及氧化等化学修饰,提供最精确“分子尺度”信息。
(8)功能活性分析:包括酶动力学、结合亲和力及细胞/ 动物模型验证,是生物功能恢复的最终证明。
生物活性检测虽是验证蛋白功能构象的重要佐证,但并非复性工艺开发的唯一终点。尤其对于缺乏明确功能指标的蛋白,其构象复性的有效性亦可由多种理化表征共同界定。构象分析的多维度评估显示,当非还原电泳与非还原高效反相色谱HPLC 分析均呈现单一条带和单峰,且荧光光谱显示出明确的构象特征指标(如色氨酸发射光谱发生特征性蓝移等)时,亦可判定蛋白已实现有效的构象复性。这一多维度表征策略能够在显著降低对功能活性检测依赖的同时,实现复性条件的高通量筛选与优化。现代复性研究正深度融合多维度试验数据与AlphaFold 等AI 工具,驱动复性工艺从传统的“经验试错”转向基于结构的“理性设计”的跨越。

06
重组蛋白药物GMP 产业化(GMP Industrialization of Recombinant Protein Drugs)
6.1 药品生产质量:GMP 产业化的核心(Pharmaceutical Manufacturing Quality:Core of GMP Industrialization)
现代化GMP 车间设计的核心围绕药品生产质量打造。以患者为中心,严格遵循中国、美国、欧盟等国家和地区及世界卫生组织(World Health Organization,WHO)等相关法规要求,深度融合“ 质量源于设计(quality by design,QbD)”与“全程风险管理”核心理念,兼顾合规性、生产效率、布局灵活性与发展可持续性。我国GMP 体系自落地后历经多轮升级,现已实现与国际法规接轨并同步吸纳国际制药工程协会(International Society for Pharmaceutical Engineering,ISPE)国际工程实践理念,通过优化生产环境,保障药品质量与稳定性,全面满足行业监管要求与实际生产需求。
6.2 质量与合规:GMP 设计的基石(Quality and Compliance: Cornerstones of GMP Design)
我国1988 年首次颁布《药品生产质量管理规范》,经1992、1998 年两次修订实现原料药与制剂全GMP 生产。2010 年修订版《药品生产质量管理规范》进一步接轨WHO 标准,融入质量风险管理理念并强化软硬件并重要求;2020 年完成生物制品附录修订,升级信息化数据采集等规范。
在设计中,通过风险评估精准识别污染源、交叉污染等各类潜在质量风险,提前采取全封闭系统等防控措施。车间布局、设备选型、工艺操作流程等核心环节经优化论证后,完成合规验证与竣工验收,建立生产全流程可追溯、质量输出一致的质量管控制度体系,为审计核查与生产持续改进提供坚实支撑,切实落实“预防为主”的GMP 管控原则。
6.3 环境控制、工艺优化与智能自动化:现代GMP 车间设计的关键(Environmental Control,Process Optimization,and Intelligent Automation :Keys to Modern GMP Facility Design)
环境控制、工艺优化与智能自动化等相关要求应契合我国《药品生产质量管理规范(2010 年修订)》标准,并同步对接ISPE 良好工程实践与高风险药品生产管控规范。
(1)环境动态管控:车间按洁净等级划分区域并设逐级过渡通道,采用高效过滤送风系统,搭配实时监测技术动态管控温湿度、压差等参数,选用易清洁耐腐蚀材质从源头防止微生物滋生。
(2)流程线性化布局:设计严守人流物流分离原则,独立设置专用通道,物料经气锁或传递窗合规进入洁净区。生产流程采用线性化布局并结合模拟工具优化,减少无效移动以适配生产需求变化。
(3)智能自动化集成:依托数字化管理系统实现生产全流程监控与数据自动记录,广泛应用智能装备减少人工操作,提升生产精度与产品一致性;智能监控系统多维度集成管理,实时分析环境及生产数据,实现设备预测性维护与远程管控。
(4)可持续发展:将可持续设计理念融入全流程,选用节能材料与设备,推行热回收、水资源循环利用,落实废弃物减量化。以绿色建筑原则降碳减排、降低运营成本,实现车间可持续发展。
当前,我国现代化GMP 车间设计以质量为核心、患者为中心,历经多轮法规修订实现与国际接轨,深度融合ISPE 工程实践理念。通过守合规、控环境、优流程,集成智能与可持续发展要求,全面适配国内外行业监管与产业发展需求。

07
重组蛋白药物质量控制理念的演进(Evolution of Quality Control Concepts for Recombinant Protein Drugs)
重组蛋白药物质量控制理念的演进,折射出生物技术产业对生命复杂性认知的不断深化。纵观其发展历程,质控理念经历了3 次重要跃迁。
第一阶段(20 世纪80 至90 年代):以“纯度即质量”为核心认知,针对大肠杆菌表达的小分子蛋白(如1982 年上市的重组人胰岛素),质控策略聚焦于分子量均一性与工艺杂质去除,主要采用SDS-PAGE、SEC 及酶联免疫吸附测定(enzyme-linked immunosorbent assay,ELISA) 等方法进行纯度鉴定和HCP、残留DNA 的总量控制[195]。然而,早期表达系统的局限,如大肠杆菌的内毒素清除与二硫键错配问题,以及酵母系统的高甘露糖型糖基化免疫原性风险等,在单纯纯度导向的质控框架下未能充分解决。
第二阶段(21 世纪00 年代):伴随单克隆抗体产业化,质控理念转向构建“微观异质性图谱”。研究人员认识到,重组蛋白是翻译后修饰(post-translational modifications,PTMs) 微观不均一的分子家族,电荷异质性、糖基化谱、氧化异构体等质量属性进入质控视野[196]。毛细管电泳(capillary electrophoresis,CE) 和MS 技术的应用使电荷异质性解析和糖基化精确定量成为可能,Fc 段岩藻糖含量对抗体依赖的细胞介导的细胞毒性作用(antibody-dependent cell-mediated cytotoxicity,ADCC)效应的影响等发现,推动质控理念从“末端检验”向“属性描述”过渡。
第三阶段(2010 年至今):以QbD 为核心理念,将质控前移至工艺设计阶段,通过关键质量属性(critical quality attributes,CQAs) 与关键工艺参数(critical process parameters,CPPs)之间的定量关系研究建立设计空间,从而实现对杂质谱的深度解析与主动调控[197-198]。在此阶段,HCP 检测从ELISA 总量控制走向质谱介导的“清单式”管理,多属性方法(multi-attribute method,MAM)结合机器学习正推动质量控制迈向“参数放行”时代[199]。
重组蛋白药物从纯度检测到微观异质性表征,再到全面的质量控制,每一次跃升都是对“何为质量”这一根本问题的重新回答。在这一历程中,分析技术是认知的工具,监管科学是规范的框架,而对临床安全与疗效的守护,则是贯穿始终的核心。

08
重组蛋白药物的非临床研究(Preclinical Research of Recombinant Protein Drugs)
重组蛋白药物的非临床研究在药学研发与首次人体试验(first-in-human,FIH)之间承担着承上启下的枢纽功能,历经40 余年演进,实现了从小分子药物模式到生物制品特色独立框架的转型[200-202]。中国该领域起步较晚但发展迅速,2007 年1 月1 日起我国要求新药非临床安全性研究必须在GLP 认证实验室进行,2017 年加入国际人用药品注册技术协调会(International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use,ICH),标志着我国非临床研究评价标准实现与国际全面接轨[203]。
当前重组蛋白药物的非临床评价遵循以ICH《S6(R1):生物制品的临床前安全性评价》[S6(R1): Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals] 为核心的国际法规框架,S6(R1)确立了“科学驱动、个案处理”的原则,已被美国FDA、欧洲药品管理局(European Medicines Agency,EMA)、日本药品医疗器械综合机构(Pharmaceuticals and Medical Devices Agency,PMDA) 及中国国家药品监督管理局(National Medical Products Administration,NMPA)全面采纳[204]。各监管机构结合自身实践形成多层次法规体系:美国FDA 于2023 年5 月发布了《药物非临床普遍接受的科学知识指导原则草案》(Draft Guidance for Industry,Generally Accepted Scientific Knowledge in Applications for Drug and Biological Products: Nonclinical Information), 允许在特定条件下利用已有知识替代部分非临床研究。近年,我国药品监管部门密集发布指导原则,如2023 年发布《抗体偶联药物非临床研究技术指导原则》等。研究策略必须紧密围绕药物的作用机制(mechanism of action,MoA)和生物学特性展开,包括相关动物种属的选择(需通过体外结合力测定、细胞功能实验及组织交叉反应性研究验证)、免疫原性评估(抗药抗体检测数据主要用于解释毒性结果,严禁直接外推预测人体风险)以及替代分子策略( 需严格的结构、功能及PK 特征桥接论证)[205]。核心研究内容涵盖重复给药毒性研究[ 识别毒性靶器官、确定未观察到有害效应的水平(no observed adverse effect level,NOAEL)]、PK 研究[ 关注靶点介导的药物清除(target-mediated drug disposition,TMDD)的非线性特征] 以及起始剂量选择 [ 高活性分子优先采用最低预期生物效应水平(minimum anticipated biological effect level, MABEL)方法][206-207]。随着蛋白药物形式多样化,非临床评价面临新的挑战:双特异性抗体需重点关注细胞因子释放综合征(cytokine release syndrome,CRS) 风险[208] ;抗体偶联药物(antibody-drug conjugate,ADC)则需融合抗体和小分子两类评价逻辑,重点考察连接子稳定性、游离毒素暴露量及靶向/ 脱靶毒性区分[209-211]。研究体系历经数十年发展,已形成以确证性试验为核心、以风险管理为保障的科学框架。

09
重组蛋白药物的临床研究进展(Advances in Clinical Research of Recombinant Protein Drugs)
现代临床评价体系由4 起历史事件塑造:1937 年磺胺酏剂事件确立上市前安全性审查[212];1942 年黄热病疫苗事件推动生物制品独立监管[213] ;1961 年沙利度胺事件促成了强制性有效性验证要求[201,214] ;20 世纪80 年代艾滋运动则开启了加速批准路径,并确定了以患者中心的临床评价模式[215]。
重组蛋白药物自1982 年重组人胰岛素上市以来, 面临PK / 药效学(Pharmacodynamics,PD)复杂性、免疫原性及种属特异性挑战。EPO诱发纯红细胞再生障碍(pure red cell aplasia,PRCA)性贫血事件推动免疫原性监测转向前瞻控制[216]。临床药理学已成为监管核心,约92%生物制品上市申请包含人体PK 数据[217-218]。模型引导的药物开发(model-informed drug development,MIDD)生理药动学(physiologically based pharmacokinetics,PBPK) 模型预测组织分布, 利用群体药动学(population pharmacokinetics,PopPK)模型量化个体间变异,并借助暴露- 效应(exposure-response,E-R)模型实现剂量优化[218-222]。
不同模态药物遵循差异化策略:单克隆抗体确立“一药多适应症”范式;生物类似药遵循逐步递进的相似性评价;双特异性抗体(bispecific antibody,BsAb) 需管控CRS 风险并遵循MABEL 原则;ADC 则需平衡靶向性、连接子稳定性与载荷毒性[208]。临床试验设计持续优化:Ⅰ 期采用贝叶斯最优区间(bayesian optimal interval,BOIN)或改良毒性概率间隔(modified toxicity probability interval,mTPI) 等基于模型的设计提高效率[223-224] ;Ⅱ期运用多重对比与建模(multiple comparison procedures and modeling,MCP-Mod)方法量化剂量- 反应关系[225-226] ;Ⅲ期确证性研究依据优效或非劣效设计。免疫原性风险分层管理将蛋白分为3 类[216],特殊人群评价需结合分子量特征。加速批准制度虽惠及患者,但43% 的抗肿瘤药需5 年以上确证获益[227-228],风险管理计划已成为高风险药物准入模板[229]。未来,真实世界证据、MIDD 及以患者为中心的开发将推动临床研究向更高效、精准的方向演进[230]。

10
重组蛋白药物的审评、审批与监管(Review,Approval,and Regulation of Recombinant Protein Drugs)
10.1 我国药物审评审批体系的演进(Evolution of China's Drug Review and Approval System)
我国药物审评审批体系在持续深化的药品监管改革中逐步完善,生物制品的审批制度经历了从“集中管理”到“科学分级”的演进历程。1999 年《新生物制品审批办法》确立了国家统一审批制度;随着2015 年《国务院关于改革药品医疗器械审评审批制度的意见》的发布、2019 年修订的《中华人民共和国药品管理法》(以下简称《药品管理法》)的实施以及2020 年版《药品注册管理办法》的颁布,我国药品监管模式已成功转型为以全生命周期管理为导向的现代化体系。
在审评理念上,该体系强调安全性、有效性与质量可控性的三位一体评估。在审批流程上,实行基于风险的分类管理,通过突破性治疗药物、附条件批准、优先审评审批及特别审批程序等,为临床急需药品提供技术指导与缩短时限等政策支持。此外,体系建立了覆盖研发全过程的沟通机制,申请人在临床试验申请前(pre-investigational new drug application,Pre-IND)、试验过程中及上市申请前(pre-new drug application,Pre-NDA)等关键节点,均可就重大技术问题与国家药品监督管理局药品审评中心(Center for Drug Evaluation,CDE)进行沟通交流,有效提升了研发效率与审评质量[231]。
10.2 上市前审评:技术准入的核心(Pre-Market Review: Core of Technical Access)
上市前审评是监管链条中技术要求最密集、专业评估最深入的环节。其核心目标是通过对药学、非临床和临床研究的全面审查,建立贯穿产品全生命周期的质量基准[232]。
根据现行药品注册管理制度,审评流程分为以下关键阶段。
(1)临床试验申请:申请人在完成非临床药学和药理毒理学研究后提交。CDE 自受理之日起60 个工作日内作出决定;对于符合要求的创新药,可在30 个工作日内完成审评审批[233]。
(2)上市许可申请:临床试验完成后,申请人提交上市申请。经技术审评、现场核查及注册检验合格后,方可获得药品注册证书。CDE 的技术审评自受理之日起200 个工作日内作出决定;对于符合优先审评审批程序的创新药,审评时限缩短至130 个工作日。
为规范重组蛋白药物的研发,CDE 发布了多项技术指导原则,深入贯彻 QbD 理念。监管要求建立覆盖生产用原材料、工艺开发、中间过程控制及稳定性研究的全过程质量控制体系,为科学研发提供明确的技术指引。
10.3 上市后监管:变更管理与动态控制(Post-Market Oversight: Variation Management and Dynamic Control)
随着法规的完善,已上市生物制品的药学变更已从“按事项管理”转向“基于风险的管理”[234]。《药品注册管理办法》明确规定,药品注册证书有效期内药品上市许可持有人应当持续保证上市药品的安全性、有效性和质量可控性。
(1)分类管理模式:2021 年施行的《药品上市后变更管理办法( 试行)》及配套指导原则,将生物制品药学变更按影响程度划分为重大、中等、微小三类。药品上市许可持有人作为责任主体,需结合产品特点开展充分的可比性研究,确保技术进步或工艺优化不影响产品质量[235-236]。
(2)日常监督检查:依据《药品生产监督管理办法》,监管部门遵循风险管理与全程管控原则,将“药物警戒制度”的实施情况纳入重点检查内容。通过建立药品安全信用档案,对有不良记录的企业增加检查频次,实施联合惩戒[237]。
对于重组蛋白药物这类复杂生物制品,全生命周期管理意味着从细胞库建立到上市后工艺变更的每一个环节,均处于科学评估之下。
10.4 小结(Summary)
综上所述,我国重组蛋白药物的审评审批与监管体系已形成科学的管理闭环。制度上以《药品管理法》为基石、《药品注册管理办法》为核心,构建了上市前后有机衔接的法规框架;技术上推动QbD 理念从研发向生产全流程延伸;运行上通过关键节点沟通与风险变更分类,实现了科学审评与动态监管的结合。这一体系为保障重组蛋白药物的临床获益提供了制度保障,也为产业高质量发展奠定了基石。

11
重组蛋白药物未来发展展望( Future Prospects of Recombinant Protein Therapeutics)
重组蛋白药物凭借高度的特异性、靶向性和优异的生物相容性,已成为治疗肿瘤、自身免疫性疾病、代谢性疾病及罕见病的核心力量,深刻改变了全球疾病治疗格局。当前,随着AI(如AlphaFold)技术的迭代突破与抗体类药物研发的持续创新,重组蛋白药物研发正迎来前所未有的发展机遇,朝着高效化、精准化与多元化的方向迈进。
11.1 AI(AlphaFold)深度赋能研发进程 [Deep Integration of AI(AlphaFold)in R&D]
DeepMind 旗下的AlphaFold 系列模型推动了AI 与蛋白质科学的深度融合,显著提升了靶点发现效率并优化了研发成本。目前,AlphaFold 3已实现多模态扩展,能够高精度预测蛋白与配体、核酸的复合物结构[141]。
新一代模型IsoDDE 实现了关键跨越,其核心优势在于三维图神经网络与物理约束的深度融合。该模型不仅能预测静态三维结构,还能解析分子间的动力学相互作用、精确穿透药物靶点并计算结合自由能,直接回答了新药研发中“药物如何作用”的核心科学问题。传统研发路径下,一款新药动辄耗时10~15 年、耗资数十亿美元,而AI 技术的介入正在全流程赋能。
(1)靶点发现阶段:AI 可快速挖掘多组学大数据,结合结构预测精准识别潜在靶点。对于传统实验难以解析的膜蛋白及复杂复合物,AI 大幅扩大了筛选范围并缩短了验证周期。例如,利用AlphaFold 预测EGFR T790M 突变型激酶结构,可快速定位结合口袋,为高选择性抑制剂的设计提供精确指导[238]。
(2)分子设计与优化阶段:生成式模型可实现全新蛋白药物的从头设计,或对现有分子进行结构修饰以优化其亲和力与稳定性。AlphaFold 4 级别模型已能直接输出药效团模型及结合亲和力等关键参数,将先导化合物的发现周期从数年缩短至数月。此外,AI 在预测毒性和免疫原性方面的应用,有助于提前排除高风险候选分子。2025 年起,美国FDA 等监管机构开始推动AI 驱动的非临床评价技术,逐步减少对动物实验的依赖。
11.2 抗体药物的多元化创新(Diversified Innovation in Antibody-Based Therapeutics)
抗体类药物作为蛋白药物研发的重要品种,正从经典的单克隆抗体向更具功能多样性的形式演进。
(1)ADC :通过特异性抗体与细胞毒性药物的精准偶联,ADC 实现了“靶向递送、精准杀伤”。截至2025 年,ADC 已成为全球抗肿瘤药物研发的重镇,其中中国企业在研产品占比已达全球的2/3。未来研发重点将聚焦于载荷优化与连接子稳定性的提升,并逐步向非肿瘤领域拓展[239]。
(2)BsAb :BsAb 通过双靶点结合实现协同治疗。2026 年有望迎来全球首个BsAb ADC 产品的上市。目前,中国在BsAb 领域的临床阶段产品占比已达全球的55%,展现出强劲的追赶与创新势头[239]。
此外,随着AI 技术的融入,复杂蛋白分子的三级结构与结合强度得到精准预测。使得融合蛋白、抗体片段(scFv/Fab)及长效抗体等亦发生快速迭代。同时抗体药物也进入多技术融合的新纪元。
11.3 原始创新与监管科学协同驱动下的rHSA 研发(rHSA R&D Driven by the Synergy of Original Innovation and Regulatory Science)
11.3.1 角色演变:从战时战略储备到临床精准应用(Role Evolution :From Wartime Strategic Reserves to Clinical Precision Applications)
人血清白蛋白(plasma-derived human serum albumin,pHSA)占血浆总蛋白的60%[36-38],提供了血浆约80% 的自由巯基[42-43],维持了血浆约80% 的胶体渗透压[39-41](笔者称之为“ 白蛋白688 原理”)。回顾历史,pHSA 的临床价值在第二次世界大战战伤救治中得到了关键性的验证:1939 年临床研究发现,血浆能有效替代全血救治失血性休克;1941 年珍珠港事件爆发后,Edwin J. Cohn 实验室紧急提供的早期“低温乙醇分离法”纯化的白蛋白,被首次用于救治严重烧伤士兵,取得显著成效[240]。这直接确立了白蛋白作为国家战略储备物资的地位,白蛋白的规模化生产与战时供应成为保障前线医疗救治体系的关键环节。这种从“实验性制品”向“战略物资”的转变,不仅推动了大规模白蛋白分离技术的发展,也为其后续在临床精准应用中的角色演变奠定了基础。在现代生物医药体系中,血源白蛋白与重组白蛋白正形成优势互补、协同保障的格局。血源制品作为资源驱动型药物,始终是保障公共卫生安全的重要战略物资。与此同时,rHSA 的发展代表了质量控制模式的演进,它通过高度一致的生产工艺,满足了临床路径中对分子构象均一性及批次稳定性的精细化需求,拓展了白蛋白在复杂生物医学场景下的应用维度。
11.3.2 跨学科前瞻:rHSA 作为 “健康稳态调节者”的潜力(Interdisciplinary Foresight: rHSA as a "Regulator of Health Homeostasis")
白蛋白分子构效关系的复杂性展现了其独特的生物学功能。尽管早期基于血浆置换的经验性疗法因缺乏精准分子机制而效果受限,但利用“年轻、无损、超纯” 重组人白蛋白(young,undamaged,and ultra-pure Recombinant Human Serum Albumin,简称PG-rHSA)调节机体稳态的策略已展现出广阔的应用前景。在小鼠模型中利用“年轻、无损、超纯” 重组小鼠白蛋白(young,undamaged,and ultrapure Recombinant Mouse Serum Albumin,简称PG-rMSA) 已取得的研究结果[45], 为PG-rHSA 的临床转化奠定了坚实基础。基于蛋白质折叠理论的演进,PG-rHSA 正成为干预系统性疾病与退行性病变的重要切入点:①衰老相关机制研究,探讨生命演化中维持健康寿命的核心通路与系统性干预手段。②神经退行性疾病,聚焦阿尔茨海默病、帕金森病的蛋白错误折叠致病机制及临床转化探索。③代谢与免疫相关疾病,从全局视角探讨 2 型糖尿病[46]、肥胖与脂代谢 [ 含笔者团队发现并命名的“白蛋白体(Albumosome)”][241] 以及类风湿关节炎等慢性炎症疾病的致病机制。④心血管系统稳态,重点关注心肌肥大、心脏纤维化等涉及心肌重构的疾病干预研究。
11.3.3 质量定义的范式转移:极限纯度与分子完整性(Paradigm Shift in Quality Definition: Ultra-Purity and Molecular Integrity)
要实现上述跨学科的预判,rHSA 的质量界定必须从传统的“纯度”向“分子水平的精准受控”转型。
(1)超高纯度(ultra-purity):目前,尖端重组技术制备的 PG-rHSA 的HCP 含量已严控在10 ng/g 以下。在质量控制实践中,笔者主张在传统的ELISA 基础上,利用其与物理表征手段的正交互补性,引入灵敏度达纳克级(≤ 1 ng)的银染 SDS-PAGE 作为关键质量属性的补充验证手段。这一策略旨在规避单一检测技术在抗体覆盖率等方面的系统性局限,通过双重验证实现对微量外源蛋白杂质更直观、全面的监控。品质评价应与其临床相关的制剂浓度(如200 mg/ml,20%)高度挂钩。在20% 的高浓度(大分子拥挤环境)下,维持分子长达两年以上的单体状态具有极高的技术挑战。稳定性研究结果验证了PG-rHSA 在该浓度及储存周期下,通过银染SDS-PAGE 检测依然保持严格的单体条带,证明该系列技术从源头上抑制了因Cys34 介导的二硫键错配,确保了空间构象的均一性。
(2)分子完整性(molecular integrity):构象均一与化学修饰的协同稳态。
除了纯度以外,重组白蛋白的高质量与否至少与以下3 项核心参数密切相关:① Cys34 位点的还原或氧化状态直接决定了白蛋白的抗氧化能力。②白蛋白的同型半胱氨酸化不但会改变白蛋白的电荷分布和空间构象,影响其对游离脂肪酸的运载功能,还会导致内皮细胞黏附力下降、炎症反应增强,并最终诱发动脉粥样硬化。③ N- 末端缺失不但使其失去结合铜离子的能力,还会导致不受控制的Fenton 反应和氧自由基的产生[242]。
11.3.4 监管科学与原始创新的协同:从质量对标到标准定义(Synergy of Regulatory Science and Original Innovation: From Quality Benchmarking to Standard Definition)
2024 年,英国政府针对20 世纪下半叶发生的血液污染事件正式发布调查报告并公开致歉,报告证实,该事件导致超过30 000 人感染HIV 或丙型肝炎病毒(hepatitis C virus,HCV),死亡人数已逾3000 人。这一全球血液制品安全史上的重大事件,再次凸显了血浆来源产品在生物安全监控与追溯体系中的极端重要性。这一全球性安全事件不仅强化了行业对血源筛选技术的研发投入,更推动了各国和地区监管机构对血液制品生产全过程风险控制方案的重新审视。早在1998 年我国实行无偿献血制度之后,通过国家药品监管部门建立的高度受控、程序规范的血浆采集与加工监管体系,有效提升了国产血浆来源产品的批次一致性与病毒安全性。
综上所述,rHSA 与pHSA 是基于不同临床路径与资源属性的战略互补。血源产品在急性扩容与基础保障中发挥着重要的支撑作用,而PG-rHSA则凭借其分子构象的高度受控与成分确定性,在精准医疗及生物制药载体领域展现出独特优势。这种由严谨监管环境与原始创新技术共同驱动的转型,正推动我国生物医药产业从单纯的质量符合向引领全球行业标准定义的维度演进。

12
结语:在硅基预测之外守护碳基尊严(Conclusion :Safeguarding Carbon-Based Dignity Beyond Silicon-Based Predictions)
自1976 年基因泰克(Genentech)在硅谷诞生,开启基因重组技术商业化元年起,重组蛋白药物研发已走过半个世纪。这五十年的辉煌,是几代人跨越时空的接力:既有深耕基础研究与临床转化的科学家、医生及无数从业者,他们从分子微观世界中破译生命的语言;也有在反应器旁枯燥打磨工艺的研发人员,将实验室的星火转化为惠及大众的产能;既有在资本寒冬中凭远见守护创新的投资人,也有在临床一线严谨把关安全的监管者;更有那群以身试药、最终受益于科学进步的患者群体。正是这种学术、产业与社会的深度协同,使我们实现了从最初简单激素的“异源表达”,到如今复杂单抗、多功能融合蛋白的代际跨越。
站在2026 年这一AI 赋能的数字化新纪元,尽管AI 驱动的“从头设计”展现了强大的分子定制与结构生成能力,预示着人类正尝试从生命密码的“被动解读者”转变为“主动书写者”,但在重组蛋白药物的临床转化中,我们仍需保持高度的科学理性。自然界从未存在过的新型蛋白,一旦作为药物进入人体,将面临严峻的免疫原性挑战。人体免疫系统对“非自然造物”的排斥,本质上是碳基生命在 38 亿年演化中,为捍卫底层生物学法则而形成的自我保护机制。因此,AI 的设计自由度绝不能脱离临床安全的底线。重组蛋白药物研发的未来,不应是毫无敬畏的“自由王国”,而必须是在遵循碳基生命免疫耐受规律的前提下,对底层科学机制的谨慎探索与精准运用。

谨以此文纪念Robert L. Baldwin教授。
This article is respectfully dedicated to the memory of Professor Robert L. Baldwin.


作者简介
罗永章,博士,清华大学生命科学学院教授,蛋白质技术国家工程研究中心主任。专业方向:衰老与抗衰老机制研究、神经退行性疾病机制研究、代谢相关性疾病机制研究以及心血管病机制研究

【参考文献】详见纸刊。




编辑:向丽
审核:赵燕宜






