 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库周末文摘 | 生物医学新技术临床研究的证据质量与证据衔接——兼论医疗机构临床研究与药械路径选择的统计学考量


引用本文
曹寒,陈晓嫒,姚晨*.生物医学新技术临床研究的证据质量与证据衔接——兼论医疗机构临床研究与药械路径选择的统计学考量[J].中国食品药品监管.2026.05(268):38-51.
生物医学新技术临床研究的证据质量与证据衔接
——兼论医疗机构临床研究与药械路径选择的统计学考量
Evidence Quality and Bridging in Clinical Research on New Biomedical Technologies: Statistical Considerations on Clinical Research in Medical Institutions and Drug/Device Pathway Selection
曹寒
清华大学北京清华长庚医院医学数据科学中心,清华大学临床医学院
CAO Han
Medical Data Science Center, Beijing Tsinghua Changgung Hospital, School of Clinical Medicine, Tsinghua University
陈晓嫒
清华大学北京清华长庚医院临床药械试验中心,清华大学临床医学院
清华大学药品监管科学研究院
CHEN Xiao-yuan
Clinical Trial Center for Drugs and Medical Devices, Beijing Tsinghua Changgung Hospital, School of Clinical Medicine, Tsinghua University
Institute for Drug Regulatory Science, Tsinghua University
姚晨*
北京大学临床医学高等研究院
海南省真实世界数据研究院
YAO Chen*
Institute of Advanced Clinical Medicine,Peking University
Hainan Institute of Real-World Data Study

摘 要 Abstract
在《生物医学新技术临床研究和临床转化应用管理条例》《生物医学新技术与药品、医疗器械界定指导原则(暂行)》以及《中华人民共和国药品管理法实施条例》陆续实施的制度背景下,本文以人脐带间充质干细胞药物艾米迈托赛注射液在激素治疗失败的急性移植物抗宿主病中的研究为例,从学术角度探讨生物医学新技术临床研究证据在不同路径选择下的应用边界,及其与药品、医疗器械注册临床证据要求之间的衔接条件,并结合医疗机构内开展相关临床研究时的管理要求,提出提高证据质量与证据衔接价值的统计设计要点。
Against the institutional backdrop of the successive implementation of the Regulations on the Administration of Clinical Research and Clinical Translational Application of New Biomedical Technologies, the Interim Guidelines for the Definition of New Biomedical Technologies, Drugs and Medical Devices, and the Regulations for the Implementation of the Drug Administration Law of the People's Republic of China, this paper takes the study of Amimatoside Injection, an umbilical cord mesenchymal stem cell therapy, for steroid-refractory acute graft-versus-host disease (aGVHD) as an example. From an academic perspective, the paper explores the application boundaries of clinical research evidence for novel biomedical technologies under different regulatory pathway selections, as well as the bridging convergence conditions between such evidence and the clinical evidence requirements for drug and medical device registration. Furthermore, in light of the management requirements for conducting relevant clinical research in medical institutions, the study proposes statistical design considerations aimed at improving evidence quality and enhancing the value of evidence bridging.

关键词 Key words
生物医学新技术临床研究;医疗机构临床研究;证据质量;证据衔接;目标试验模拟;ALCOA+ ;电子源数据
clinical research on new biomedical technologies; clinical research in medical institutions; evidence quality;evidence bridging; target trial emulation; ALCOA+; eSource

基金项目
昌平实验室资助课题(2023C-10-01)

生物医学新技术临床研究在未满足临床需求的探索、适应症扩展可行性评估、剂量和给药策略优化、特殊人群证据补充以及上市后再评价等方面具有重要价值。其中,研究者发起的临床研究(investigator-initiated trial,IIT)是医疗机构内开展此类研究的重要实现形式之一[1],但《生物医学新技术临床研究和临床转化应用管理条例》(国务院令第818 号,以下简称《管理条例》)[2]所规范的生物医学新技术临床研究并不完全等同于IIT。对生物医学新技术而言,研究对象往往异质性强、作用机制复杂、长期安全性和制备一致性问题突出,若希望将初步研究结果用于后续注册沟通、关键性研究设计优化,或作为支持性证据进行讨论,则研究从一开始就必须按照“证据质量与证据衔接价值”而非“仅能发表”的标准来设计。
2026 年5 月1 日起施行的《管理条例》《生物医学新技术与药品、医疗器械界定指导原则(暂行)》(以下简称《界定指导原则( 暂行)》)[3] 以及2026 年5 月15 日起施行的《中华人民共和国药品管理法实施条例》(国务院令第828 号,以下简称《实施条例》)[4],共同构成了当前讨论生物医学新技术临床研究证据用途的制度背景。《界定指导原则(暂行)》明确,临床研究发起机构应参照《生物医学新技术临床研究备案指导清单》对拟开展临床研究的生物医学新技术属性进行自主界定;对于涉及生物医学新技术与药品的界定,界定为“生物医学新技术”并拟据此开展备案临床研究的,其临床研究产生的数据用于支持生物医学新技术临床转化应用;自主界定属于药品、医疗器械(以下简称药械)的,则应按照药械管理规定依法依规开展相关工作。此外,国家卫生健康委员会(以下简称国家卫生健康委)等部门于2024 年9 月印发《医疗卫生机构开展研究者发起的临床研究管理办法》,明确医疗卫生机构开展的IIT,是指不以药械(含体外诊断试剂)等产品注册为目的的临床研究,并对医疗卫生机构主体责任、分类管理和干预性研究条件作出规定[5]。由此,生物医学新技术临床研究与IIT 之间既有交叉,也不能简单等同;讨论其证据用途时,应将技术路径、药械路径与医疗卫生机构临床研究管理要求一并纳入分析框架。
需要特别指出的是,依据《管理条例》第二十九至三十一条,生物医学新技术临床研究结束后拟转化应用于临床的,应由临床研究发起机构向国务院卫生健康部门提出申请,并由国务院卫生健康部门组织技术评估和伦理评估后作出批准或者不予批准决定。由此可见,技术路径下的“临床转化应用审批”与《实施条例》项下药械注册审批并非同一制度路径,两者在审批主体、适用对象和证据用途上均存在明确区别[2]。
基于上述制度背景,本文更侧重从学术角度讨论生物医学新技术临床研究在路径识别前提下的证据质量要求与证据衔接条件,并补充讨论其中在医疗卫生机构内开展、符合《医疗卫生机构开展研究者发起的临床研究管理办法》适用情形的项目所面临的管理与方法学问题,而非预设任何技术路径下产生的数据都可以直接进入新药审评证据链。本文结合《管理条例》《实施条例》《界定指导原则(暂行)》和《医疗卫生机构开展研究者发起的临床研究管理办法》的制度要点,并以人脐带间充质干细胞(mesenchymalstem cells,MSC)药物艾米迈托赛注射液证据链为例,探讨这类临床研究在何种前提下可用于支持生物医学新技术临床转化应用、后续药械路径沟通、关键性研究设计优化以及支持性证据评价,并提出一套适用于证据衔接要求的统计设计要点。
1《界定指导原则(暂行)》与双条例实施后的制度边界及证据衔接
1.1《 管理条例》重塑了生物医学新技术临床研究的前端门槛
《管理条例》将“生物医学新技术”界定为以对健康状态作出判断或者预防治疗疾病、促进健康为目的,运用生物学原理,作用于人体细胞、分子水平,在我国境内尚未应用于临床的医学专业手段和措施。该文件要求,在开展生物医学新技术临床研究前,应当依法完成实验室研究、动物实验等非临床研究,并由非临床研究证明该技术安全、有效后方可进入临床研究。这意味着,无充分非临床研究基础的技术探索性临床研究将面临更高制度风险。
同时,《管理条例》把研究机构条件、治理结构和备案要求前移到研究启动阶段:实施机构原则上应为三级甲等医疗机构,须具备符合要求的学术委员会和伦理委员会、稳定经费来源、相应设施与研究能力;研究项目必须经学术审查和伦理审查通过,并在5 个工作日内完成备案,备案材料中应包含研究工作基础、临床研究方案、风险预防控制措施、应急处置预案、知情同意书样式和经费使用方案等。换言之,研究方案不再只是学术论文,而是受监管核查的制度性文件。
更值得关注的是实施环节的连续义务。《管理条例》要求研究必须按备案方案实施;涉及研究目的、研究方法、主要研究终点、统计方法和受试者等的实质性变更,需重新经学术和伦理审查,并在5 个工作日内变更备案;受试者书面知情同意、严重不良反应暂停与伦理再评估、定期报告、研究结束后的长期随访、原始记录至少保存30 年等要求,均提示生物医学新技术临床研究必须按照可核查、可追责的逻辑运转。
1.2《 实施条例》将“药品研制”要求前移到研究全过程
与《管理条例》相呼应,《实施条例》进一步强调,从事药品研制活动,应当遵守药物非临床研究质量管理规范、药物临床试验质量管理规范,保证记录和数据真实、准确、完整和可追溯。这一表述直接把很多过去被视为“数据管理层面”的要求,上升为药品研制活动的法定义务。
《实施条例》还明确了药物临床试验申办者的核心责任:选择具备相应能力的药物临床试验机构和研究者,并履行受试者保护、临床试验用药品管理、临床试验数据管理和风险管理等职责;试验用药品制备应符合药品生产质量管理规范的有关要求;申办者和机构不得向受试者收取与临床试验有关的费用。由此可见,一旦研究的本质目的转向支持药品上市许可,监管关注点将不再停留于“临床技术有没有希望”,而是进一步延伸到“该证据链是否满足药品审评所要求的全流程质量控制”。
1.3 路径识别:如何区分临床转化应用路径与药械路径并完成前端选择
《管理条例》第五十五条提出,生物医学新技术与药械的界定指导原则由国家卫生健康委会同国家药品监督管理局根据科学技术的发展进行制定和调整;《界定指导原则(暂行)》则进一步把这一要求具体化,明确临床研究发起机构应参照《生物医学新技术临床研究备案指导清单》对拟开展临床研究的生物医学新技术属性进行自主界定[3]。其制度含义在于:对于处于临床前研究和临床研究等早期阶段、创新性强、个性化程度高且尚未或难以开发为药械的生物医学新技术,可以按照技术路径开展备案临床研究;而对自主界定属于药械的研究对象,则应按照药械管理规定依法依规开展相关工作。
因此,实践中的关键并不是笼统地讨论研究中途是否需要转向药械注册,而是在研究启动前结合研究对象属性、产品化程度和证据用途,完成技术路径与药械路径的前端选择。下列情形通常提示应优先考虑药械路径:其一,研究对象具有明确的产品形态、标准化制备工艺、批次放行和质量标准要求;其二,研究目的已不再主要是技术可行性探索,而是拟用于支持特定适应症、给药方案、关键终点或关键性研究设计;其三,研究者或发起方已经预期将研究结果用于药械注册沟通或者上市许可申请中的有效性、安全性和风险- 获益判断。相反,对于个性化程度高、尚未形成同类机制原理成熟药械路径且更适合作为前期探索和临床验证的新技术,则可在《界定指导原则(暂行)》框架下沿技术路径开展研究。对于其中由医疗机构组织实施且不以药械产品注册为目的的相关临床研究,还应同时纳入《医疗卫生机构开展研究者发起的临床研究管理办法》所规定的机构主体责任、分类管理和全过程质量管理要求[5]。
从制度设计看,《管理条例》已经为生物医学新技术设置了相对独立的“ 临床研究- 临床转化应用” 管理链条。尤其是第二十九至三十一条明确:临床研究结束后拟转化应用于临床的,应由临床研究发起机构向国务院卫生健康部门提出申请,提交临床研究报告和记录、适用范围、不良反应和禁忌、应用机构和人员条件、临床应用操作规范以及风险预防控制措施等材料;国务院卫生健康部门受理后,将申请资料转交专业机构开展技术评估和伦理评估,并据此作出批准或者不予批准决定。换言之,技术路径的后续目标首先是临床转化应用审批,而非当然进入药械注册审批程序[2]。
综上所述,更稳妥的表述是:技术路径下备案临床研究形成的数据,原则上用于支持生物医学新技术临床转化应用;药械路径下形成的证据,则应按照药械注册管理要求组织和表达。两种路径既不能混同,也不宜重复。对于在医疗机构内开展且不以药械产品注册为目的的相关临床研究,还应遵循国家卫生健康委关于IIT 的管理要求。对希望将临床研究结果用于后续药械研发讨论的研究而言,前提不是事后“补注册语言”,而是在启动时就完成边界识别、路径选择,并据此确定相应的质量体系、比较框架和证据表达方式。
1.4 生物医学新技术临床研究初步证据的评价框架
基于《管理条例》《实施条例》和《界定指导原则(暂行)》的衔接要求,生物医学新技术临床研究初步证据是否具有后续证据衔接价值,可概括为4 个连续判断:法律属性是否明确、研究用途是否清楚、研究设计是否支撑因果解释、数据质量是否达到可核查水平。生物医学新技术临床研究初步证据质量与证据衔接价值的四重判断框架,详见表1。

2 从“可发表”到“可用于证据衔接”:提高生物医学新技术临床研究证据质量的方案设计要点
2.1 先写清监管问题和证据用途,而不是先做研究后找注册场景
面向后续证据衔接的生物医学新技术临床研究,首先要在方案首页或研究背景部分明确“该研究究竟要回答什么决策问题”,即研究是用于适应症探索、剂量优化、给药路径选择、目标人群富集、关键终点筛选、外部对照可行性验证,还是用于补充长期安全性信息。不同用途决定不同设计要求,也决定结果在后续沟通和评价中的权重。
如果研究者预期将此类临床研究证据用于后续药品监管沟通,方案中不宜仅写“探索性观察”或“评价安全性和有效性”,而应进一步写明其在未来证据衔接中的定位,如为关键性研究提供目标人群收窄依据、为单臂设计提供比较锚点、为后续真实世界外部对照建立变量字典和结局定义,或为长期随访方案提供事件监测框架等。只有证据用途预先明确,后续方案、数据和统计解释才可能形成一条完整链条。
2.2 随机化仍是优先设计,单臂关键性研究必须以目标试验模拟为框架
随机对照试验在混杂控制上具有不可替代的优势,在具备随机实施条件时,仍应作为确证疗效的优先设计。对于危重症、罕见病、无标准治疗或伦理上难以开展随机化的场景,单臂研究、外部对照与长期随访可作为补充路径,但此类设计并非无需严谨对照,而是必须通过科学设计证明其能够回答监管所关注的因果问题。单臂设计应尽可能贴近一项可明确阐释的“目标试验”,外部对照也不可在研究结束后临时选取数据库,而应在研究启动阶段同步完成数据可用性评价、变量映射、终点判定一致性、时间零点对齐及缺失数据模式评估[6]。因此,在无法开展随机化的生物医学新技术临床研究中, 唯有基于目标试验模拟(target trial emulation,TTE) 框架开展规范设计,依托外部对照的单臂研究结果才具备被监管机构接受与讨论的基础。
TTE 是将观察性数据转化为可靠因果证据的核心方法,其核心思想是将观察性分析严格设计为对一项假设随机对照试验的严谨模拟,从而系统性消除永生时间偏倚、选择偏倚与错分类偏倚。基于提升健康研究质量与透明度(Enhancing the Quality and Transparency of Health Research,EQUATOR)国际协作网络制定的《目标试验模拟观察性研究的透明报告规范》(Transparent Reporting of Observational Studies Emulating a Target Trial,TARGET)[7],为TTE 观察性研究提供了全流程透明报告的统一规范。其要求研究者先明确定义理想目标试验方案,再将各要素逐项映射至观察数据,并完整报告因果假设、分析细节与偏倚控制措施,使监管机构能够清晰地评价设计合理性与证据可信度。在无法开展随机对照试验的生物医学新技术临床研究中,遵循 TARGET 规范实施TTE,是实现稳健因果推断并获得监管认可的关键路径。实施TTE 时,研究者需首先完整构建目标试验的核心要素,包括明确的入排标准、待比较的治疗策略、随机分配规则、统一时间零点、规范随访周期、清晰临床终点及待估计的因果对比效应,并明确条件可交换性、正值性与一致性等因果识别假设。在此基础上,需将目标试验各要素精准转化为观察数据的可操作定义,包括目标人群、入排标准、时间零点、基线窗口、治疗策略、允许合并用药、比较策略、主要终点、随访窗口、估计目标,以及提前停药、救援治疗、死亡、失访等伴发事件的处理规则与混杂控制策略。目标试验到观察数据的完整映射必须在方案阶段预先明确,而不是在分析阶段回溯构建, 这也是 TARGET规范提升研究透明度与可重复性的核心要求[8]。
从监管审查角度看, 基于TARGET 规范的TTE 设计需满足以下关键要点:①关注时间零点定义是否统一合理、能否有效避免永生时间偏倚,这是观察性因果分析最常见的设计缺陷。其次,治疗策略需清晰可操作,避免模糊的暴露/ 非暴露分组,明确启动、剂量调整、中断、重启及终止规则,这直接决定结果的可解释性。②因果估量需与监管问题高度契合,与 国际人用药品注册技术协调会(International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use,ICH)《E9(R1):临床试验中的估计目标与敏感性分析》[E9(R1): Addendum on Estimands and Sensitivity Analysis in Clinical Trials] 估计目标框架保持一致,明确区分意向治疗效应与符合方案效应,这是审评判断证据相关性的重要依据。③因果识别假设的陈述与论证、对未测量混杂的审慎讨论,以及基于不同假设、操作定义与统计模型的全面敏感性分析,均为监管评估证据稳健性的核心内容。④ TARGET 规范还要求完整地报告受试者筛选流程、组间基线特征、缺失数据分布、效应估计与精度及全部敏感性分析结果,使审评人员可完整复现研究逻辑并系统评估偏倚风险。
2.3 终点、估计目标与伴发事件处理应直接服务于审评判断
面向后续审评讨论与证据衔接的生物医学新技术临床研究,其终点设定不能仅以数据易获取性或统计敏感性为导向,而必须优先选择与临床获益直接相关、能够支撑风险- 获益判断、定义清晰且可重复量化评价的指标,确保研究结果能够直接回应药械审评所关注的核心问题。对于细胞治疗、基因治疗等先进治疗产品,由于其作用机制复杂、起效周期长且潜在远期安全性风险突出,在终点设计时还需结合疾病自然史、产品作用特点与长期风险特征,前瞻性预设中长期疗效结局与长期安全性监测指标,避免终点选择过窄或随访周期不足导致证据链无法满足审评需求。
估计目标策略[9] 是连接研究设计、统计分析与审评决策的关键框架,能够有效避免研究执行、数据解读与研究初衷脱节。在方案阶段必须明确界定本次研究拟估计的真实治疗效应,清晰地区分是“既定治疗策略下的总体治疗效果”,还是“剔除救援治疗等干扰因素后的方案依从人群效果”,并与 ICH E9(R1) 估计目标框架[10] 保持一致。在此基础上,需对临床实践中常见的伴发事件制定标准化处理规则,包括救援治疗启动、提前停药、交叉使用其他干预措施、死亡、失访、方案偏离等情形,明确主分析策略与敏感性分析路径,确保不同情境下的统计推断均具备稳健性与可解释性。
对于用于注册沟通、支持审评决策的生物医学新技术临床研究而言,终点选择、估计目标界定与伴发事件处理并非单纯的统计技术问题,而是直接决定证据可信度的核心设计要素。完整、规范且预先明确的设定,不仅能够提升研究的科学性与严谨性,更是提高此类研究证据质量并支持后续证据衔接的必要前提。
2.4 统计分析坚持“主分析清楚、扩展分析透明、稳健性可验证”
统计分析计划(statistic alanalysis plan,SAP)应在研究开始前明确主要和次要终点、估计目标、分析集、缺失数据处理、方案偏离处理、亚组分析、中期安全性审查及敏感性分析。对创新方法的使用,建议坚持“主分析简明清楚、扩展分析用于稳健性验证”的原则,避免把复杂模型作为唯一证据来源。在小样本或单臂场景中,倾向评分方法、逆概率加权、层级模型和贝叶斯外部信息借用可以提高估计效率,但其解释价值高度依赖于外部数据来源是否透明、借用强度是否可控、敏感性分析是否充分。因此,方案中应同时预设“如果外部借用假设不成立,主结论是否仍可解释”的稳健性检验路径。
国家药品监督管理局药品审评中心在相关指导原则中鼓励采用创新统计方法,但强调其应用必须基于充分的科学依据,且分析结果应具备稳健性[11-12]。在样本量有限、缺失数据较多或需整合多源信息的场景下,传统频率论方法常面临统计效能不足、模型假设过强等局限,而将贝叶斯统计与传统方法融合,可为上述问题提供可行的解决路径[13-15]。贝叶斯方法的核心价值在于能够将历史研究、外部数据或先验知识量化为先验分布,并纳入当前分析框架,从而在控制混杂的基础上提升估计效率与推断可靠性[16]。例如,倾向性评分与贝叶斯融合模型,可先通过倾向性评分平衡观测混杂,再在均衡数据层内开展贝叶斯分析[17-18] ;也可将倾向性评分直接作为协变量纳入贝叶斯多因素回归[19-20],同时对关键变量赋予信息性先验,实现混杂控制与先验信息利用。在面向后续审评讨论的相关临床研究中,这类融合方法多作为敏感性分析策略,用于验证主要结论在不同统计假设下的稳定性[11,16]。
2.5 将产品工艺一致性和给药可重复性写入方案,而不是留到化学、生产与质量控制阶段再补
对细胞治疗、基因治疗以及其他高度依赖制备工艺的生物医学技术而言,研究结果能否被用于后续监管评价,往往不仅取决于临床终点本身,也取决于研究对象是否可以被视为同一产品。方案中应明确产品来源、关键原材料、制备工艺版本、批次放行标准、储运条件、给药前准备、给药操作窗口和偏差容忍范围,并建立关键工艺变更的记录与评估机制。
如果研究期间发生制备工艺、质控标准、批次释放规则、储运条件或给药方式变化, 必须在方案和统计解释中说明这些变化对可比性和外推性的影响。对希望与药械监管证据要求形成衔接的生物医学新技术临床研究而言, 产品一致性不是药学团队的“附属问题”,而是影响临床可解释性的核心变量。
3 伦理与合规:双条例衔接下不可回避的底线要求
3.1 学术审查、伦理审查、备案与注册沟通应同步设计
《管理条例》要求生物医学新技术临床研究必须经学术审查和伦理审查通过后方可开展,并在规定时间内完成备案。《界定指导原则(暂行)》进一步明确,临床研究发起机构应参照《生物医学新技术临床研究备案指导清单》自主界定属性,并科学合理选择技术路径或药械路径。与此同时,《医疗卫生机构开展研究者发起的临床研究管理办法》明确,医疗卫生机构是相关临床研究实施的责任主体,并应建立健全组织体系、质量体系、利益冲突防范机制和研究参与者权益保护机制[5]。因此,对拟产生较高证据质量并服务后续证据衔接的生物医学新技术临床研究而言,研究者不宜把备案视为纯行政程序,而应借此同步完成研究方案、知情同意书、风险管理计划、数据管理计划、产品管理要求以及路径选择之间的一致性校核;若项目同时符合《医疗卫生机构开展研究者发起的临床研究管理办法》的适用情形,还应满足其全流程管理要求。此外,此类研究不能出现“研究已经开始,规则仍在形成”的局面。若研究者已预期未来可能转向药械路径沟通,则宜在正式入组前完成研究问题、主要终点、统计方法、外部对照策略和长期随访方案的稳定化。
3.2 书面知情同意与受试者保护不是形式要件,而是设计质量的一部分
《管理条例》要求取得受试者或者其监护人的书面知情同意,并以受试者或者其监护人容易理解的方式告知其研究目的、研究方案、可能风险和受试者权益,不得以欺骗、胁迫或利诱方式取得同意;《实施条例》亦强调申办者对受试者保护承担责任。由此可见,知情同意不是研究完成后的补充手续,而是决定研究是否具备较高证据质量和后续证据衔接价值的基础条件。对危重症、代理同意、长期随访或可能重复给药的场景,方案应预设补充同意、再次同意或撤回同意后的数据处理规则。若方案变更可能影响受试者权益,应重新获取书面知情同意。建议研究者为此保留完整的版本控制、签署留痕和培训记录。
目前临床研究的数字化转型[21]深刻改变了受试者权益保护的实践场景, 其中电子知情同意(electronic informed consent,eIC)的应用是核心变革[22-23]。eIC 并非简单的纸质流程电子化,而是需要同时满足法律合规、伦理要求与技术可靠的综合管理体系。法律层面,eIC 应符合《中华人民共和国电子签名法》可靠电子签名规定,并遵循《中华人民共和国个人信息保护法》对敏感健康信息的单独同意与数据安全要求。伦理层面,eIC 可通过多媒体展示、互动确认等方式提升受试者理解程度,动态同意模式更能保障研究全过程的自主选择权。监管审查重点关注 eIC系统的安全性、知情过程的实质有效性以及弱势群体的可及性,同时要求在推广电子化模式的同时保留必要的线下辅助渠道,确保伦理覆盖的完整性。
3.3 风险预防、暂停/ 终止规则与长期随访义务必须前移写入方案
《管理条例》要求研究机构采取风险预防、控制和处置措施;发生严重不良反应时,应暂停研究并由伦理委员会评估是否继续实施;研究结束后还应对受试者进行随访监测,评价长期安全性和有效性。对细胞和基因治疗相关产品而言,这些要求与药品审评对远期风险监测的关注高度一致。
因此,研究方案中应写清暂停标准、终止标准、严重不良事件和特殊关注事件的判定与报告流程、独立数据监查委员会(Data and Safety Monitoring Board,DSMB)或同类安全监测机制,以及研究结束后的随访频次、随访窗口和关键长期结局。
3.4 隐私保护、数据最小化与跨系统流转管理
生物医学新技术临床研究通常涉及电子病历(electronic medical record,EMR)、实验室信息系统(laboratory information system,LIS)、影像归档与通信系统(picture archiving and communication system,PACS)、随访平台、生物样本系统和研究数据库之间的数据流转,若再叠加外部对照或多中心数据整合,个人信息保护和跨系统一致性风险会显著上升。研究者应在方案、知情同意书和数据管理计划中清晰地界定数据采集范围、去标识化策略、访问权限、传输加密、委托处理关系、日志留痕和备份恢复机制。
4 以ALCOA+ 和可追溯为核心的数据质量体系
无论是《管理条例》关于及时、准确、完整记录并留存原始材料的要求,还是《实施条例》关于“记录和数据真实、准确、完整和可追溯”的要求[4],其底层逻辑都可以归结为ALCOA+,即可归属性(attributable)、可读性(legible)、同步性(contemporaneous)、原始性(original)、准确性(accurate),以及完整性(complete)、一致性(consistent)、持久性(enduring)、可用性(available)。对拟进入后续审评与沟通证据链的生物医学新技术临床研究而言,ALCOA+ 是方案设计、研究实施、统计解释和现场核查都要落地的质量框架。ALCOA+ 在生物医学新技术临床研究中的落地要点与常见缺陷,详见表2。

4.1 源数据采集、系统验证与审计追踪要在研究启动前闭环
研究者应明确哪些数据直接来源于EMR、实验室、影像、受试者报告结局或研究现场记录,哪些属于研究专属采集数据,哪些经过接口导入、映射或衍生进入电子病例报告表(electroniccase report form,eCRF)和分析数据库。对于电子源数据系统、电子知情同意平台、随机系统和研究数据库, 应保留适用性验证、权限配置、时间同步、备份恢复、版本变更和审计追踪记录。当前生物医学新技术临床研究的数据管理正从传统纸质病例报告表的分散模式,向基于电子源数据(eSource)的智能化、一体化模式转型;其中在医疗机构内开展、符合《医疗卫生机构开展研究者发起的临床研究管理办法》适用情形的项目,更应结合机构全流程质量管理要求落实数据治理。电子源数据通过直接从临床原始记录中采集数据,避免二次转录,从源头保障数据的真实性、完整性与可追溯性[24-26]。在高质量证据研究框架下,电子源数据系统需与医院信息系统(hospital information system,HIS)、LIS、PACS 深度集成, 实现数据自动或半自动填充,最大限度减少人工干预,提升数据质量与合规水平。
4.2 外部对照和真实世界数据的质量门槛必须追溯到数据形成机制
当生物医学新技术临床研究拟使用外部真实世界数据时,审评关注点绝不仅是样本量,而更看重数据形成机制是否与研究问题匹配。研究者至少应说明外部数据的采集场景、纳入规则、关键变量来源、缺失模式、结局判定方式、数据抽取与清洗规则、冻结时间以及与前瞻性研究时间零点的对齐策略。对于无法追溯关键变量来源、无法确认治疗起始时间或无法统一结局定义的数据库,不宜作为主要比较来源。构建高质量真实世界数据平台是开展外部对照研究的重要前提,平台需遵循标准化、高质量、安全合规与系统互操作原则,采用通用数据模型实现多源异构数据整合治理,形成研究型数据集[27]。
4.3 从源数据到分析数据集的质量控制应形成可核查的数据流
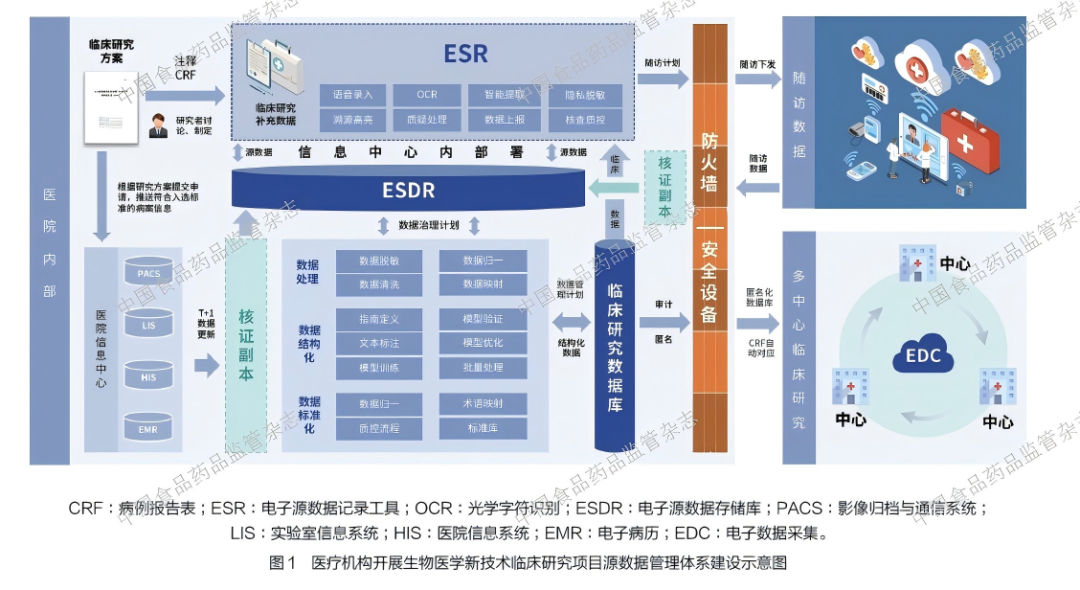
高质量生物医学新技术临床研究数据管理计划应展示完整数据流:源数据采集、质疑处理、逻辑核查、中央监查、外部接口导入、编码、医学审核、统计程序生成分析集、锁库和归档。对关键终点、安全性变量、入排标准判断变量、产品批次和给药过程变量,应设置更高频次的核查要求[28]。在此基础上,端到端(End-to-End,E2E)全过程数据管理理念进一步推动了全周期数据质控体系建设,依托一体化电子源数据存储库(electronic source data repository,ESDR)[28]可实现院内系统部署与验证,完成数据采集、确认、前置质控与自动化流转,显著提升数据采集的标准化、及时性与准确性,强化数据留痕、核查与全生命周期追踪能力,为此类研究及细胞治疗类研究提供可复制的源数据驱动管理框架。
4.4 数据治理体系与伦理合规实施
在数据治理与合规实施层面,临床研究可以依托ESDR 开展全流程数据管理,实现与HIS、LIS、PACS 及EMR 的深度集成,形成从源数据采集、质控、清洗、分析到归档的E2E 数据管理体系,如图1 所示。所有临床数据、实验室检查、影像学评估与疗效终点均通过电子源数据直接捕获,避免二次转录误差,并依托系统内置的逻辑核查、范围核查与审计追踪功能,实现数据实时质控与全流程可追溯,满足 ALCOA +与监管核查要求。同时,本课题研究全面采用eIC 开展受试者权益保护,通过可靠电子签名保障签署过程的真实性、完整性与不可篡改性,借助多媒体展示、分层告知与动态同意机制提升知情理解度与受试者自主权,在危重症与多中心场景下, 既保障伦理合规性,又提升研究实施效率与远程执行能力,充分体现数字化转型在真实世界研究中的合规价值。
5 案例解读:从现有研究看提高证据质量与证据衔接价值的关键要素
5.1 已发表Ⅱ期随机对照结果对目标人群富集和关键性研究设计的启示
2025 年1 月, 首款MSC治疗药物艾米迈托赛注射液(商品名睿铂生, 药物研究代号hUC-MSC PLEB001)经国家药品监督管理局附条件批准上市,适应症为治疗14 岁以上消化道受累为主的激素治疗失败的急性移植物抗宿主病(acutegraft-versus-host disease,aGVHD)。本文以该MSC 药物为例,分析其从探索性研究到Ⅱ期随机对照试验,再到后续关键性研究设计优化的证据链,并从证据衔接和路径选择角度进行讨论。已发表的Ⅱ期结果研究[29] 为多中心、随机、双盲、安慰剂对照探索性Ⅱ期试验,共纳入78 例受试者,其中MSC 组40 例、对照组38 例;在各中心自选一种二线治疗(不含芦可替尼)的基础上比较MSC 与安慰剂的疗效与安全性;主要评价指标为第28 天总体缓解率(overall response rate,ORR)。意向性治疗分析(intention-to-treat analysis,ITT)结果显示,第28 天 MSC组与对照组的ORR 分别为60.0% 和 50.0%(P=0.375),差异无统计学意义。但是,符合方案分析(per-protocol analysis,PP)结果中,MSC 组与对照组第28 天 ORR 分别为 71.9% 和46.7%(P=0.043), 差异具有统计学意义;特别是在消化道受累亚组中,MSC 组的疗效显著优于对照组(第28 天 ORR 分别为66.7% 和 33.3%,P=0.031)。此外,安全性分析发现两组不良事件发生率总体相近且未观察到输注相关毒性。
在上述Ⅱ期试验结果基础上,后续向国家药品监督管理局药品审评中心申请Ⅲ期关键性临床试验时,并未简单沿用全人群随机对照逻辑,而是转向“聚焦消化道受累人群的单臂关键性研究”路径。该多中心单臂关键性临床研究现已正式发表[30] :共纳入54 例受试者, 第28 天ORR 为63.0%(95%CI :48.7%~75.7%),完全缓解率为55.6% ;第56 天ORR为51.9%, 治疗总体耐受性良好,未观察到输注相关毒性或治疗相关严重不良事件。其核心并不是回避对照,而是借鉴TTE 的设计思想,在前瞻性单臂研究中尽可能前移处理未来影响监管判断的偏倚问题:第一,以Ⅱ ~ Ⅳ度消化道受累的激素治疗失败aGVHD 患者作为目标人群,减少由异质性过大带来的选择性偏倚;第二,以首次启动研究给药作为统一时间零点,固定第28天ORR 为主要终点,统一访视与判定窗口,降低信息偏倚;第三,为所有受试者规定统一的背景二线治疗策略(以抗CD25 单抗为主),并将新增第二种系统性二线治疗界定为治疗失败,从而尽量控制联合治疗差异带来的混杂偏倚;第四,预先设定客观效能标准,以既往研究和历史对照信息形成单臂研究的客观绩效标准(objective performance criterion,OPC), 增强结果可解释性。
5.2 证据衔接视角下的启示
该案例的探索性Ⅱ期试验证据与药品Ⅲ期注册证据要求的衔接,不是靠事后为阳性结果寻找解释,而是依赖“探索性随机对照试验发现信号- 目标人群富集-关键性研究前瞻性验证- 监管批准”的连续证据递进。其一,Ⅱ期随机对照试验的ITT 主分析未达显著,提示探索性研究本身不能被简单外推为确证性证据;其二,消化道受累亚组人群中MSC组第28 天 ORR 相比安慰剂呈现显著优势,为后续关键性研究提供了科学的人群富集依据;其三,Ⅲ期单臂关键性研究通过TTE 框架统一背景治疗、明确时间零点、预设OPC 阈值和标准化失败定义,提高了单臂证据的可解释性与可核查性;其四,最终获批的适应症并非笼统的激素治疗失败的aGVHD, 而是收窄至14 岁以上消化道受累为主的激素治疗失败 的aGVHD,这说明后续证据采用更看重目标人群界定是否充分、证据链是否递进一致。由此可见,真正提高证据质量与证据衔接价值的关键,不是简单强调研究“有阳性结果”,而是把目标试验范式、对照选择逻辑、数据核查路径、产品质量控制和统计稳健性前移设计,使证据结构、偏倚控制与后续评价要点形成闭环。
6 生物医学新技术临床研究证据与药械注册证据要求衔接的实践清单
为增强可操作性,本文提出如下实践清单。对拟将此类临床研究证据用于后续路径选择、关键性研究设计优化或支持性证据评价的研究,建议至少形成下列表单和动作闭环,详见表3。

7 结论
在《管理条例》《实施条例》和《界定指导原则(暂行)》共同构成的制度背景下,生物医学新技术临床研究证据具有与药械证据要求形成衔接的可能,但这种“可衔接性”绝不是自发产生的。其前提是研究对象法律属性明确,研究用途与拟解决的问题一致,研究设计能够控制关键偏倚,产品和给药过程具有可比性,伦理与受试者保护安排合规,数据链条真实、准确、完整、可追溯并经得起核查。
因此,真正面向证据衔接的生物医学新技术临床研究,不宜作为注册临床试验的直接替代,也不宜在论文表述上预设为“进入新药/ 械审评证据链”。更符合《界定指导原则(暂行)》和《管理条例》第二十九至三十一条的表述是:技术路径下形成的研究数据主要用于支持生物医学新技术临床转化应用,且其后续临床转化应用审批由国务院卫生健康部门负责;若研究对象属性、产品形态和证据用途已更符合药械研发,则应及早选择相应药械路径。对于其中在医疗机构内开展,且不以药械产品注册为目的的相关临床研究,还应同步遵循国家卫生健康委关于IIT 的管理要求。对研究者而言,关键不在于研究结束后再为结果寻找路径语言,而在于在方案阶段就完成从临床问题、法律属性、路径选择、研究设计、受试者保护、产品一致性、源数据治理到统计解释的系统化布局。只有这样,生物医学新技术临床研究才能从“可发表的早期探索”逐步转化为“可被后续讨论和参考的前端证据”。
第一作者简介
曹寒,博士,清华大学北京清华长庚医院医学数据科学中心,清华大学临床医学院,助理研究员。专业方向:临床研究方法学
通讯作者简介
姚晨,教授,临床研究方法学博士生导师,北京大学临床医学高等研究院临床研究所副所长,海南省真实世界数据研究院副院长。专业方向: 临床研究统计设计与分析
参考文献:详见纸刊。




编辑:李丹
审核:赵燕宜






