五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库ASCO:一个靶点正在崛起
2024年3月26日,日本厚生劳动省(MHLW)批准了VYLOY(
zolbetuximab
)用于治疗CLDN18.2阳性、不可切除、晚期或复发性胃癌患者。这是
世界上第一个
针对
CLDN18.2
阳性,不可切除,晚期或复发性
胃癌
患者的CLDN18.2靶向治疗,也是唯一一个获得世界监管机构批准的CLDN18.2靶向疗法,目前该疗法已经在国内获批上市。
zolbetuximab的获批已经证实了该靶点的成药性,并且已经多种机制的药物进入临床,在今年的ASCO会议上,包括TCE,ADC,以及PD-L1,CD47 ,4-1BB在内的多款药物披露了临床疗效(
文末对比数据汇总
)。
QLS31905 是一款靶向 Claudin18.2/CD3 的双特异性抗体。在今年的ASCO会议上,齐鲁将会公
QLS31905 联合化疗一线治疗 Claudin18.2 阳性胰腺癌(PC)和胃癌(GC)
的安全性与疗效数据。
方法:这项 Ib/II 期试验招募了 Claudin 18.2 阳性(定义为
1% 的肿瘤细胞染色强度
1+)、局部晚期不可切除或转移性胰腺癌和胃癌患者,且既往未接受过系统性抗肿瘤治疗。
队列 1(胰腺癌)
:接受 QLS31905(350 ug/kg、500 ug/kg 或 800 ug/kg,每 2 周或每 3 周一次)联合吉西他滨+白蛋白紫杉醇治疗。
队列 2(胃癌)
:接受 QLS31905(500 ug/kg 或 800 ug/kg,每 3 周一次)联合奥沙利铂+卡培他滨治疗。
结果:
截至 2025 年 12 月 11 日,共入组 88 例胰腺癌患者和 43 例胃癌患者。
安全性
:
未发生剂量限制性毒性(DLT),未达到 MTD
。最终确定的 RP2D 为
800 ug/kg 每 3 周一次
(适用于胰腺癌和胃癌)。队列 1 和队列 2 中,分别有 71.6%(63例)和 60.5%(26例)的患者发生了≥3 级治疗相关不良事件(
TRAE
)。因 TRAE 导致停药的比例分别为 11.4%(10例)和 9.3%(4例)。两个队列均未发生 QLS31905 相关的死亡事件。
疗效(胰腺癌,82 例可评估)
:
ORR 为 59.8%
(95% CI: 48.34%-70.44%),
DCR 为 89.0%
。中位无进展生存期(PFS)为
8.74 个月
,中位缓解持续时间(DoR)为 8.94 个月,中位总生存期(OS)为
15.87 个月
。
低表达亚组
(15例)
:即便在 Claudin18.2 低表达患者中,ORR 仍达 60.0%,DCR 达 93.3%,中位 OS 为 15.61 个月。
疗效(胃癌,43 例可评估)
:
ORR 高达 74.4%
(95% CI: 58.83%-86.48%),
DCR 为 93.0%
。中位 PFS 为 10.09 个
月,中位 DoR 尚未达到。
低表达亚组(9例
)
:
ORR 为 77.8%,DCR 为 100%
,9 个月 PFS 率为 83.33%。
结论:QLS31905 联合化疗用于 Claudin18.2 阳性胰腺癌和胃癌的一线治疗,展现出
可控的安全性
和
极具潜力的疗效
。目前,III 期临床试验正在进行中,以进一步确证其在胰腺癌患者中的疗效与安全性。

再看
启愈生物
Q-1802,其是
靶向 CLDN18.2 和 PD-L1双特异抗体。Q-1802 一方面可通过 Fc 段介导抗体依赖性细胞毒性(ADCC)和吞噬作用(ADCP)直接杀伤表达 CLDN18.2 的肿瘤细胞;另一方面通过阻断 PD-1/PD-L1 结合激活免疫系统以消除肿瘤。
在今年的ASCO会议上,
启愈生物将披露
Q-1802 联合 XELOX 一线治疗 CLDN18.2 阳性晚期胃癌/胃食管结合部癌(GC/GEJ)的临床疗效。

Qure-1802-201 是一项多中心、开放标签、非随机的 I/II 期临床试验。
入组标准
:经组织学确认的
不可切除局部晚期/转移性 GC/GEJ,HER2 阴性
,且既往未接受过治疗;CLDN18.2 阳性定义为 ≥40% 的肿瘤细胞呈现 2+/3+ 膜染色。治疗方案:患者接受 Q-1802(
10 mg/kg 或 20 mg/kg,每 2 周一次)联合标准 XELOX 化疗
(奥沙利铂 d1 静滴;卡培他滨 d1-14 口服,每 3 周一次)。
截至 2025 年 12 月 11 日,共入组 62 例患者(10 mg/kg 组 45 例,20 mg/kg 组 17 例)。
安全性方面
:未观察到剂量限制性毒性(DLT),未达到 MTD。所有患者均发生治疗期不良事件(TEAE),Q-1802 相关 TEAE 主要包括恶心(75.8%)、AST 升高(61.3%)、ALT 升高(51.6%)和疲劳(50.0%)。≥3 级 TEAE 和 ≥3 级 Q-1802 相关 TEAE 的发生率分别为 74.2% 和 43.5%。最常见的 ≥3 级相关不良事件为血小板减少(8.1%),其次为中性粒细胞减少(6.5%)。因 TEAE 导致永久停药的比例为 6.5%,无治疗相关死亡。
疗效方面(60 例可评估)
:
ORR 高达 70.0%
(42/60,95% CI: 56.8%–81.2%),
疾病控制率(DCR)接近 100%
(仅 1 例未获益)。中位无进展生存期(mPFS)为
11.3 个月
(95% CI: 8.0–16.8)。
生物标志物分析
:疗效与 CLDN18.2 表达呈正相关。在 CLDN18.2 高表达(2+/3+ ≥70%)患者中(10 mg/kg 组,N=37),ORR 为 73.0%,mPFS 为 11.3 个月。在上述基础上,若合并 PD-L1 CPS ≥5(N=11),疗效进一步提升:ORR 达 81.8%,mPFS 延长至 12.2 个月。20 mg/kg 组的 ORR(70.6%)与 10 mg/kg 组(69.8%)相似,提示 10 mg/kg 可能为最佳剂量。

恒瑞的SHR-3821 治疗晚期实体瘤患者的 I 期临床研究也会在今年的ASCO会议上披露,不同于前两个药物,
SHR-3821 是一款 ADCC 增强型双特异性抗体,可特异性靶向 CLDN18.2,并以 CLDN18.2 依赖的方式激活 4-1BB。其临床
入组患者为 CLDN18.2 阳性、
对标准治疗无响应或缺乏标准治疗的晚期实体瘤患者
。

截至 2025 年 11 月 30 日,共 40 名 CLDN18.2 阳性晚期实体瘤患者(23 例胃癌 GC,17 例胰腺癌 PC)接受了 0.1 至 20 mg/kg 剂量的 SHR-3821 治疗。
安全性:未发生剂量限制性毒性(DLT),在整个评估剂量范围内未达到 MTD。15 名患者(37.5%)发生了 ≥3 级治疗相关不良事件(TRAE),最常见的是中性粒细胞计数减少(20.0%)、
淋巴细胞计数减少
(7.5%)和呕吐(5.0%)。无 TRAE 导致死亡,仅 1 名患者(2.5%)因 TRAE 停药。药代动力学(PK):初步 PK 分析显示,单次给药 1–20 mg/kg 后,全身暴露量随剂量成比例增加,末端消除半衰期(t1/2)范围为4.2 至 7.0 天。
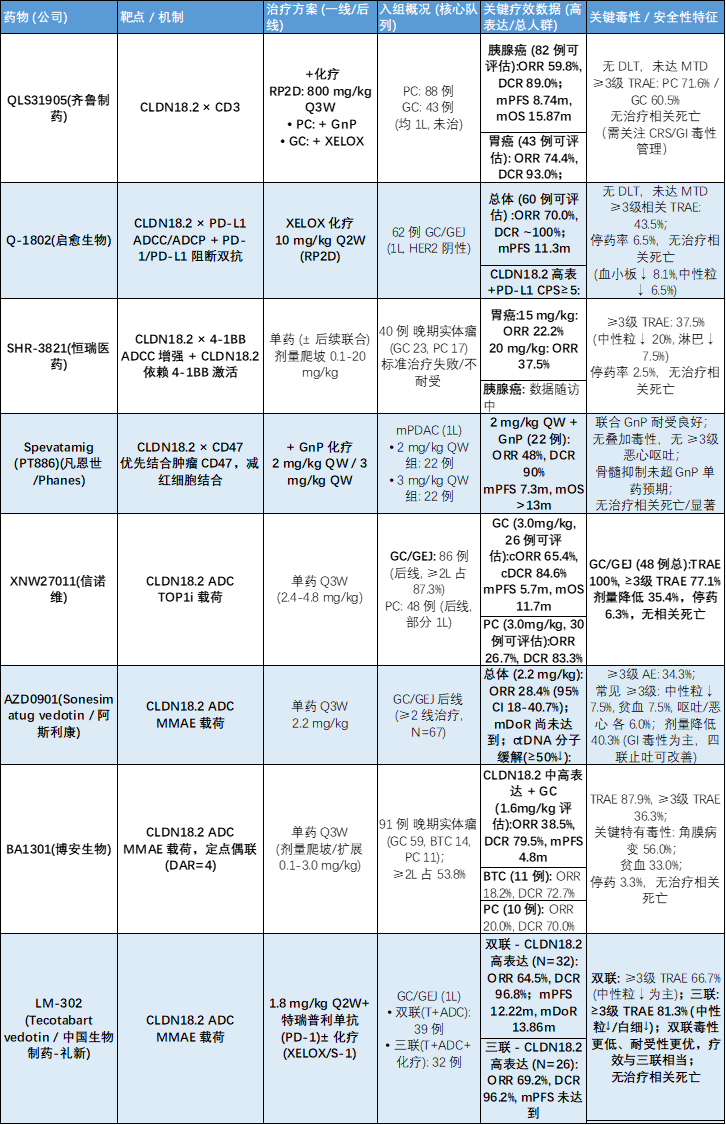
疗效方面
:胃癌(GC)
:15 mg/kg 组 ORR 为
22.2%
,20 mg/kg 组 ORR 提升至
37.5%
。
凡恩世的PT886是
靶向 CLDN18.2 和 CD47 的双特异性抗体。其抗 CD47 臂经过优化设计,能优先结合癌细胞表面的 CD47,而非人红细胞表面的 CD47(从而降低血液毒性)。本次ASCO会议上
凡恩世将披露
PT886
联合吉西他滨+白蛋白紫杉醇(GnP)一线治疗转移性胰腺导管腺癌(mPDAC)的数据。

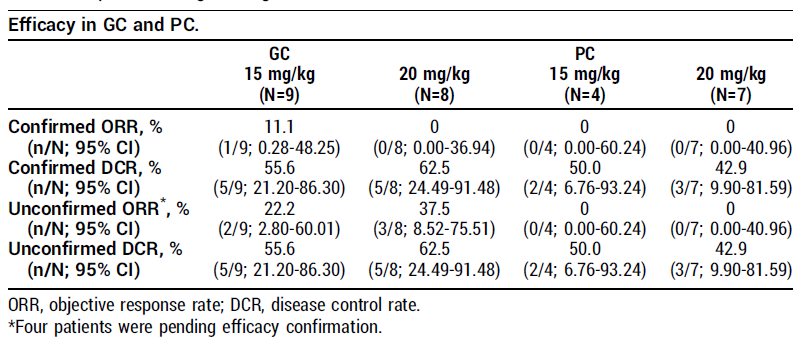
这次的临床研究报告将聚焦于 II 期队列:Spevatamig 联合 GnP 一线治疗 mPDAC,包含两个剂量组:2 mg/kg QW(N=22)和 3 mg/kg QW(N=22)。
截至 2026 年 1 月 15 日,共有 151 名患者在单药或联合治疗背景下接受了 Spevatamig 治疗。
2 mg/kg QW + GnP 组(N=22):
安全性:Spevatamig 联合化疗耐受性良好。骨髓抑制(血细胞减少)发生率未超过 GnP 单药预期水平;无 ≥3 级恶心或呕吐事件。
疗效方面
:
ORR 为 48%
,
DCR 为 90%
。中位 PFS(mPFS)为
7.3 个月
(95% CI: 5.6 个月 – 尚未达到);中位 OS(mOS)
超过 13 个月
(数据尚不成熟)。中位随访时间为 8.9 个月。
生物标志物
:无论肿瘤 CLDN18.2 表达水平如何,也无论是否存在 RAS 突变,均观察到了抗肿瘤活性。
3 mg/kg QW + GnP 组(N=22)
:安全性方面,
在现有安全数据中,与单独使用 GnP 的预期不良事件谱相比,
未发现显著的叠加毒性
;
无
3 级恶心或呕吐事件
。该组的最新详细安全数据将在会议上公布。

当然,除了TCE等双抗,
CLDN18.2
ADC药物
的临床疗效也不错,即将举办的ASCO会议上多个ADC公布了临床疗效,包括
信诺维的XNW27011,
阿斯利康的
AZD0901,
博安生物的BA1301,
中国生物制药的LM-302
。
首先是
信诺维的XNW27011
,本次会议上
信诺维将公布
NW27011 治疗 CLDN18.2 阳性胃癌/胃食管结合部腺癌(G/GEJA)患者 II 期研究数据和
后线治疗
胰腺癌(PDAC)的
II 期研究数据
。
首先是治疗
G/GEJA的数据,
这项 II 期研究旨在评估 XNW27011 的疗效和安全性。
入组标准
:患有
晚期实体瘤、且对标准治疗失败/不耐受/无可用标准治疗
的成年患者,要求
CLDN18.2 表达为 IHC ≥2+ 且阳性肿瘤细胞 ≥5%
(依据 EPR19202 标准),其中包括 G/GEJA 患者。所有患者均有可测量病灶(RECIST v1.1)且 ECOG 评分为 0-1。
给药方案
:患者接受 XNW27011(2.4-4.8 mg/kg,每 3 周一次 Q3W)。
结果:截至 2025 年 12 月 29 日,共入组 86 例符合条件的 G/GEJA 患者(剂量范围 2.4-4.8 mg/kg)。研究选定
IHC
≥
2+、TC
≥
20%
作为 XNW27011 后续开发的 CLDN18.2 表达标准。以下报告为该标准下的患者数据:
安全性(71 例患者)
:中位年龄 57.0 岁;87.3% 的患者既往接受过
≥
2 线系统治疗;85.9% 的患者既往接受过免疫检查点抑制剂。常见的(
≥
20%)治疗相关不良事件(TRAE)包括:贫血、恶心、白细胞减少、食欲下降、中性粒细胞减少、呕吐、体重减轻、低白蛋白血症、乏力、血小板减少、淋巴细胞减少和低钾血症。
疗效(3.0 mg/kg 剂量组,26 例可评估)
:患者基线:23 例接受过
≥
2 线治疗,3 例接受过 1 线治疗。中位随访时间(mFU)为 11.3 个月。
确认的客观缓解率(cORR)为 65.4%
,确认的疾病控制率(cDCR)为
84.6%
。
中位无进展生存期(
mPFS
)为
5.7 个月
,中位总生存期(mOS)为
11.7 个月
。
亚组分析
:在 23 例接受过
≥
2 线治疗的患者中,mPFS 延长至
6.8 个月
,mOS 为
11.9 个月
。
值得注意的是
,一名既往曾接受过其他 CLDN18.2 靶向治疗的患者仍表现出持久的临床获益,PFS 已达 11.3 个月,且截至数据截止仍在继续治疗。

胰腺癌(PDAC)的
II
期研究数据:
治疗入组标准
是CLDN18.2 阳性(定义为 ≥5% 肿瘤细胞 IHC 染色 ≥2+)且既往接受过系统治疗的 PDAC 患者。
给药方案
:接受 XNW27011(2.4 - 4.8 mg/kg,每 3 周一次 Q3W)。
截至 2025 年 12 月 29 日,共入组 48 例患者,其中大部分入组 3.0 mg/kg 剂量组。
患者基线
:
ECOG PS
为 0 或 1,中位年龄 59.0 岁。18 例(37.5%)既往接受过 1 线系统治疗,22 例(45.8%)既往曾接受过拓扑异构酶 I 抑制剂(TOP1i)治疗。
疗效(3.0 mg/kg 组,30 例可评估)
:ORR 为 26.7%
,
DCR 为 83.3%
。中位 PFS(mPFS)为
4.1 个月
,中位 OS(mOS)为
10.0 个月
。1 线经治亚组(13 例)
:ORR 显著提升至
46.2%
,DCR 达到
100.0%
,mPFS 为 4.4 个月。虽然 OS 尚未成熟,但在中位随访 8.1 个月时,当前 mOS 已达
10.1 个月
。既往接受过 TOP1i 亚组
:在中位随访 7.9 个月时,mPFS 为 5.2 个月,mOS 为 10.0 个月(显示出对既往化疗耐药患者依然有效)。
安全性(48 例)
:治疗相关不良事件(TRAE)发生率为
100.0%
。≥
3 级 TRAE 发生率为
77.1%
,严重 TRAE(SAE)发生率为
58.3%
。35.4% 的患者因 TRAE 导致剂量降低,6.3% 导致停药。最常见的 TRAE 包括:恶心(77.1%)、呕吐(64.6%)、食欲下降(64.6%)和贫血(62.5%)。

再来看一下阿斯利康的
AZD0901(
Sone-Ve
),
单药治疗 Claudin 18.2 阳性(CLDN18.2+)晚期或转移性胃/胃食管结合部(GEJ)癌症患者(CLARITY-PanTumor01 研究的数据)。
CLARITY-PanTumor01
是一项正在进行的全球多中心 II 期研究(NCT06219941),旨在评估 Sone-Ve(
抗 CLDN18.2 抗体与MMAE
) 在中国以外地区(包括胃癌/GEJ、PDAC 和 BTC)的疗效。在今年的ASCO会议上,将聚焦于 子研究 1
:针对 CLDN18.2 阳性、晚期或转移性胃/GEJ 癌且既往接受过
2 线系统治疗的患者。给药方案
:患者接受 Sone-Ve 2.2 mg/kg 静脉输注,每 3 周一次(Q3W)。(1.8 mg/kg 随机组的数据未在此报告)。
截至 2025 年 10 月 31 日,共有 67 例患者接受了 Sone-Ve 2.2 mg/kg 治疗,分别来自随机队列(RC,30 例)、配对活检队列(PBC,31 例)和日本安全性队列(JSC,6 例)。
安全性
:所有患者均发生不良事件(AE);总体
≥
3 级 AE 发生率为 34.3%(RC 组 36.7%,PBC 组 35.5%)。最常见的
≥
3 级 AE 包括:中性粒细胞计数减少(7.5%)、贫(7.5%)、呕吐(6.0%)和恶心(6.0%)。剂量调整
:总体 40.3% 的患者因不良事件导致剂量降低(RC 组高达 56.7%,PBC 组为19.4%)。
胃肠道(GI)毒性
是剂量降低的最主要原因。
疗效
:
ORR 为 28.4%
(95% CI: 18.0%–40.7%)。
中位缓解持续时(DoR)
尚未达到
。
分子缓解(ctDNA)
:
在可评估的患者中,58.6%(17/29)基线 ctDNA 水平足够用于分析。
在这 17 例患者中,
10 例(58.8%)
在开始Sone-Ve 治疗 2 个周期内,ctDNA 水平降低了至少 50%(达到部分分子缓解)。
接着是
博安生物的BA1301
,
BA1301 是一款由全人源抗 CLDN18.2 单克隆抗体通过
位点特异性糖基偶联(site-specific glycol conjugation)
技术,与强效细胞毒药物单甲基澳瑞他汀 E(MMAE)连接而成的 ADC。其药物抗体比(DAR)为 4,并采用可裂解连接子。此次ASCO会议上报告 BA1301 的 I 期剂量递增和扩展研究结果。
截至 2025 年 12 月 22 日
,共入组 91 例晚期癌症患者(胃癌 59 例 [64.8%],胆道癌 14 例 [15.4%],胰腺癌 11 例 [12.1%] 及其他)。
基线
:49 例(53.8%)患者既往接
受过 ≥2 线系统治疗
(中位 2 线,范围 1-7)。
安全性(全体,N=91)
:80 例(87.9%)发生至少 1 起治疗相关不良事件(TRAE),33 例(36.3%)发生 3 或 4 级 TRAE。最常见的 TRAE
:
角膜病变(Corneal disorder,56.0%)
和贫血(33.0%)。3 例(3.3%)因 TRAE 停药,
无治疗相关死亡
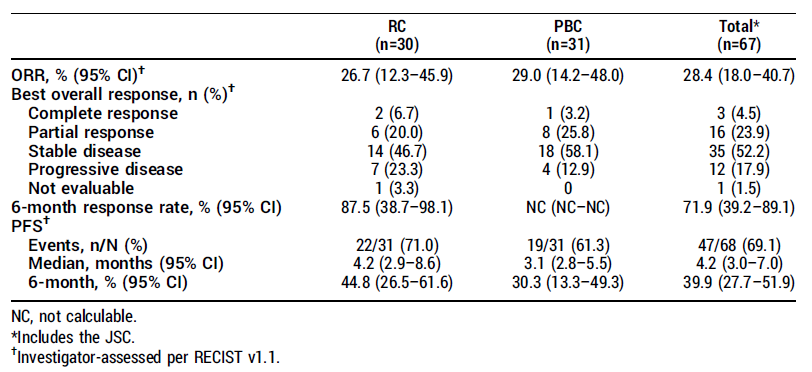
。疗效(CLDN18.2 中高表达患者,N=76)
:在 1.6 mg/kg 剂量水平(这是目前看来较优的剂量),69 例患者有基线后的肿瘤评估数据。
关键疗效数据如下表所示
:
接下来是中国生物制药的LM-302(
Tecotabart vedotin) 联合特瑞普利单抗(Toripalimab)± 化疗一线治疗 Claudin18.2 阳性晚期胃/胃食管结合部腺癌的疗效与安全性:II 期研究结果。
截止
2025 年 11 月 25 日
共入组 71 例患者:双联组 39 例,三联组 32 例。
基线特征
:两组中分别有 82.1% 和 84.4% 的患者为 Claudin18.2 高表达(≥25%);PD-L1 CPS ≥1 的患者比例分别为 61.5% 和 81.3%。
疗效(全人群)
:中位随访时间分别约为 16 个月(双联)和 15 个月(三联)。
中位 PFS
:双联组
10.68 个月
,三联组
12.55 个月
。两组的中位总生存期(OS)均
尚未达到
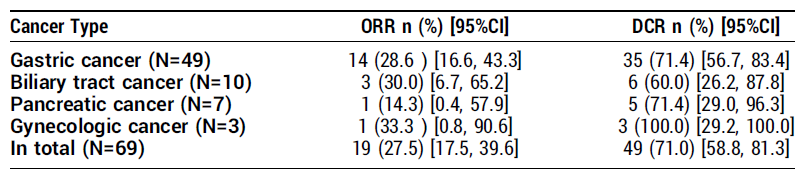
。疗效(Claudin18.2 高表达亚组,N=52)
:
双联组
:ORR
64.5%
,DCR
96.8%
,中位 DOR
13.86 个月
,中位 PFS
12.22 个月
。
三联组
:ORR
69.2%
,DCR
96.2%
,中位 PFS
尚未达到
(NE)。
Claudin18.2 低表达亚组,N=12)
:双联组(N=7):ORR 14.3%,DCR 71.4%,中位 PFS 5.72 个月。三联组(N=5):ORR 40.0%,DCR 100%,中位 PFS 6.80 个月。