 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库自动化设备漏检被FDA点名的思考!
对于注射剂视觉检查,美国FDA曾发出多封警告信,直指药品生产基地的注射剂生产存在严重违反当前良好生产规范(CGMP)的行为。其中,自动化视觉检查设备未充分检出颗粒等缺陷、长期未开展100%人工复检、CAPA程序失效等问题,成为核心违规点。
今天,我们结合FDA警告信的原文违规细节,深度拆解指南核心要求,帮你把自动化验证从“合规达标”做到“质量可控”。
一、警告信直击核心问题,戳中行业常见盲区
如FDA在2023年1月对Baxter Healthcare Corporation的检查中发现,百特的注射剂生产存在多项致命漏洞,警告信原文对违规行为的描述精准且严厉,这些问题恰恰是药企在自动化视觉检查中最易忽视的关键点:
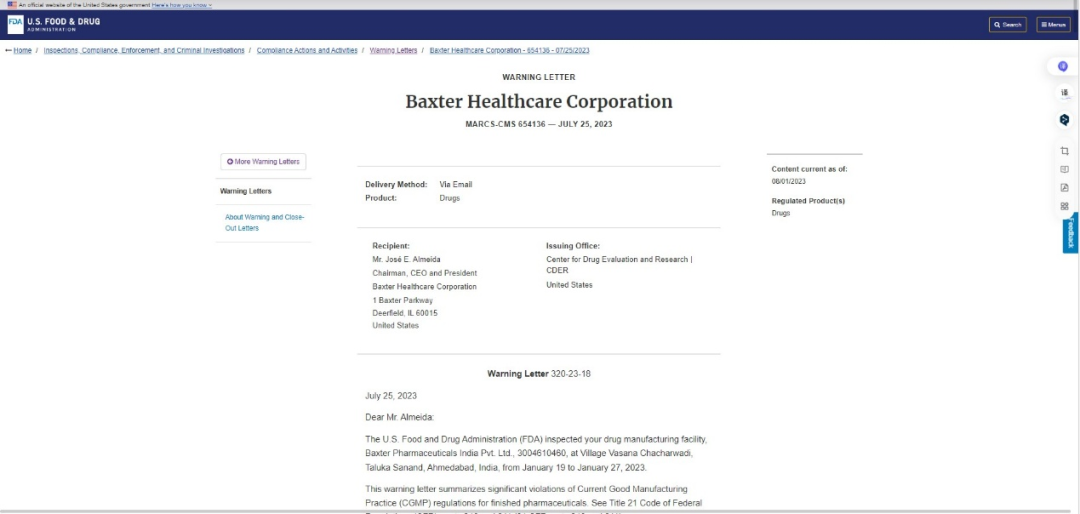
1.自动化设备“带病上岗”,缺陷检出形同虚设
FDA警告信指出:贵司未充分核查自动检测仪漏检注射剂颗粒物等已知缺陷的失效问题,虽数据证实该设备缺陷检出能力严重不足,仍持续用其检测多批次上市注射剂。
2022年10月百特缺陷挑战试验佐证设备存在重大短板:试验刻意剔除检出难度更高的颗粒,仅采用易检出的颗粒开展测试,依旧暴露设备检出性能缺陷。
企业全程单一依靠该自动化设备,未实施全量人工目检复核,无法保障经该设备检测的无菌注射剂无可见异物
2混淆“自动化”与“替代人工”,违背合规本质
FDA警告信明确:自动异物检测仅作00%人工目检的补充,设备性能需充分验证,且自动化不能替代人工检查容器裂纹等其他缺陷。百特却单用该自动化设备,违背此项CGMP合规要求。
二、ECA V5指南拆解:自动化验证的“全流程合规手册”
FDA的警告,本质上是对药企“自动化设备验证不到位、流程管控缺失”的警示。而ECA V5指南恰好为解决这些问题提供了清晰路径——从“设备行不行”到“用得好不好”,从“一次性验证”到“持续可控”,每一步都有明确的原文依据和实操标准。
1.先厘清2个核心概念:资质认定(Qualification)vs验证(Validation)
资质认定是证明设备“适用于预定用途”的书面证据,而验证是证明设备在生产过程中“持续产出符合质量要求的可重复结果”的书面证据。
2.三级资质认定:从“装得对”到“能达标”的层层把关
自动化设备的资质认定必须经过IQ、OQ、PQ三级验证,缺一不可,每一级都有明确的原文要求:
(1)IQ(安装确认):基础不牢,地动山摇
实操落地重点:
硬件一致性:相机数量、光源类型、输送轨道规格等需与URS完全匹配,例如“全视角覆盖”需确认相机安装角度无盲区;
环境适配性:安装环境需满足温度18-25℃、湿度40%-60%,避免相机受潮或光源衰减;
文档完整性:收集供应商提供的设备手册、校准证书、维护计划等,确保后续运维有据可依。
(2)OQ(运行确认):功能稳定是前提
实操落地重点:
权限控制:操作员无参数修改权限,管理员操作全程留痕,符合数据完整性要求;
输送稳定性:测试不同剂型(如5ml西林瓶、10ml注射器)的输送速度波动≤5%,无卡顿、倾倒现象;
报警与追溯:模拟输送卡顿、相机故障等场景,设备需实时报警并记录故障时间、批次信息,审计追踪数据至少保存5年。
(3)PQ(性能确认):核心中的核心,必须“人机equivalence”
PQ测试的实操落地示例
需使用“产品专属资质套件”:包含真实产品或物理/光学特性一致的模拟品,缺陷类型覆盖关键(如容器裂纹)、主要(如颗粒)、次要(如轻微划痕),确保测试场景与实际生产一致。
套件需重复检查≥10次:避免单次测试的偶然性,确保数据具有统计意义,例如某颗粒缺陷需经过10次测试均能稳定检出。
需与人机比对:安排3-5名合格人工检查员对同一套件检查,自动化设备的关键缺陷检出率需=100%,主要缺陷检出率≥人工的95%。
此外,指南原文特别强调粒子检测需遵循Knapp测试原则:Knapp证明,合格单元中缺陷单元比例不超过25%可获得最佳统计准确性;实际应用中,资质套件通常含约10%缺陷单元,以避免误判偏差。
实操中需注意:缺陷比例过高会让设备“过度敏感”,过低则无法有效验证检出能力,10%是兼顾准确性与实用性的最优比例。
3.验证(Validation):从“能达标”到“持续达标”
资质认定通过后,还需通过验证证明设备在日常生产中持续稳定,验证的2种核心实施方式):
人机比对复现:选取已通过自动化检查的批次或子批次(如5000 units),由合格人工组重新100%检查,确认设备检出率与人工无差异;
多批次AQL验证:连续验证3批生产批次,确保每批AQL抽样结果符合预设limits(关键缺陷0,主要缺陷≤0.65),证明设备在规模化生产中稳定达标。
此外,验证结果需作为regulatory submission的重要数据,需明确记录设备参数、测试方法、结果分析等信息,确保可追溯。
4.再确认与再验证:避免“一验永逸”
自动化设备并非“一验永逸”,指南原文要求建立定期复核机制:
年度再确认三项工作:复盘全年设备变更、故障维修记录;统计检出与误判趋势,关键缺陷检出率降幅超3%需排查根因;使用含裂瓶、大颗粒的测试套件检测,防止参数漂移。
连续再验证:按ASTME2587-2收集每批AQL数据绘制趋势图,数据稳定受控可免频繁全项再验证,实现批次级持续确认。
日常操作管控:每批前后使用功能测试套件,长时间生产、设备异常需中间增测;套件需包含大颗粒、裂瓶、空瓶典型缺陷;套件缺陷漏检即刻停机检修,复测合格后方可生产;连续生产8小时以上,每4小时加测一次。
三、合规落地3大避坑指南,远离FDA警告
结合百特案例与ECAV5指南,药企需规避三大合规误区:
1.文档留存不全:IQ/OQ/PQ、日常测试记录需完整记录操作人员、时间、样本、结果及异常处置;自动化PQ需留存相机参数截图、人工对照原始数据。完整文档可尽早识别设备长期验证缺陷。
2.测试缺陷脱离真实生产:验证套件缺陷需取自实际产品,禁用实验室人工非标缺陷。百特试验剔除难检出颗粒,造成设备检出性能评估失真。
3.验证通用化一刀切:PQ需分产品、剂型单独执行;采用括号法验证须经质量部审批并提供科学等效依据。百特统一参数验证多款注射剂,违反产品专属PQ要求,是漏检隐患主因。
结语:
自动化不是“合规捷径”,而是“责任升级”
注射剂自动目检可提升效率与检测一致性,但不可牺牲合规与患者安全。FDA针对百特的警告信,直指设备资质不足、未调查检出缺陷等验证简化类违规,敲响合规警钟。
ECA V5指南把自动化检查验证标准化、量化、可追溯,覆盖IQ、PQ、日常测试及年度再确认全流程。
药企需按指南落地SOP,这是GMP硬性要求,也是保障患者安全的核心义务,合规无可规避。
更多内容,可关注:





