 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库中国药科大学郑禄枫:肝癌干细胞生态位的调控机制及干预路径研究进展
肝癌干细胞生态位的调控机制及干预路径研究进展 PPS
郭颖,韩雪丹,郑禄枫*
(中国药科大学生命科学与技术学院,江苏 南京 211198)
郑禄枫

中国药科大学生命科学与技术学院副教授,博士研究生导师,博士;江苏省“青蓝工程”优秀青年骨干教师培养对象,国家自然科学基金评审专家,教育部学位论文评审专家;《中国药科大学学报》《药学进展》青年编委。主要致力于肿瘤干细胞在肿瘤发生发展中的作用机制和靶向药物开发。作为通信/第一作者在J Hematol Oncol、Mol Cancer、Oncogene、J Adv Res、Adv Sci、J Med Chem等权威学术期刊上发表论文60余篇,高被引论文6篇,H指数28;授权发明专利5项。近年来先后主持多项国家级及省部级项目,包括国家自然科学基金面上项目2项、国家自然科学基金青年项目1项、中国博士后面上项目、中国博士后特别资助项目、江苏省博士后科研资助计划及中央高校重点项目等。同时参与国家重点研发计划1项、国家自然科学基金面上项目3项。
[摘要]肝癌是全球范围内发病率和死亡率均较高的恶性肿瘤,其治疗后的高复发率和高转移率是临床面临的主要挑战。肝癌干细胞(liver cancer stem cell,LCSC)被认为是驱动肝癌发生、发展、耐药、复发及转移的关键细胞亚群。肝癌干细胞并非独立存在,其干性维持、自我更新及耐药特性高度依赖于其所在的特殊微环境——LCSC 生态位。该生态位由肿瘤相关成纤维细胞、肿瘤相关巨噬细胞、调节性 T 细胞、内皮细胞等多种基质细胞,以及复杂的细胞外基质和丰富的信号分子共同构成。系统综述了 LCSC 生态位的细胞组成、关键分子调控网络,并深入探讨了靶向生态位组成细胞、靶向分子调控网络,以及诱导肝癌干细胞分化这 3 种肿瘤治疗策略。此外,总结了当前研究面临的技术与转化挑战,并对未来研究方向进行展望,旨在为开发根除 LCSC、改善肝癌患者预后的新型联合治疗模式提供理论依据和思路。
肝癌是全球癌症相关死亡的第三大原因,其 5年生存率仍然不理想,其中肝细胞癌 (hepatocellular carcinoma,HCC) 是最主要亚型[1]。近年来,肿瘤干细胞 (cancer stem cell,CSC) 理论的提出为理解肿瘤异质性、治疗抵抗和复发提供了新的视角。肝癌干细胞 (liver cancer stem cell,LCSC)是肝癌组织中一小群具有自我更新、多向分化潜能和强致瘤能力的细胞亚群,被认为是肝癌复发、转移和耐药的“种子”细胞[2]。
LCSC 的功能和命运并非由其自身完全决定,而是深受其周围微环境——干细胞生态位的调控。肿瘤干细胞生态位是一个动态的、复杂的功能性单元,由特定的基质细胞、细胞外基质 (extracellular matrix,ECM) 成分、可溶性因子以及特殊的物理化学条件 (如缺氧、低 pH) 共同构成[3]。该生态位通过提供必要的物理锚定、细胞间接触信号和旁分泌信号,精密地调控着 LCSC 的静息、活化、自我更新与分化平衡,并保护其免受化疗药物和免疫系统的攻击[4]。因此,深入解析 LCSC 生态位的组成结构与功能,阐明其内在的分子调控网络,对于开发能够特异性根除 LCSC、克服治疗抵抗的新型策略至关重要。
本文系统综述近年来在 LCSC 生态位研究领域取得的重要进展,讨论该领域当前面临的主要挑战,并对未来研究方向进行展望,以期为推动肝癌治疗模式的革新提供参考。
LCSC 生态位的功能实现,依赖于其内部高度异质性的细胞集群所构建的复杂社会性网络[5]。因此,解析生态位的细胞组成,实质上是破译其内部通信密码的第一步。本节将聚焦于这一网络中最关键的几类细胞,包括肿瘤相关成纤维细胞 (cancerassociated fibroblast, CAF)、 肿 瘤 相 关 巨 噬 细 胞(tumor-associated macrophage,TAM)、调节性 T 细胞(regulatory T cell,Treg)以及内皮细胞,逐一阐述它们在生态位中的具体作用及其与 LCSC 的核心交互机制。
1.1 肿瘤相关成纤维细胞与肝癌干细胞的交互作用
CAF 是肿瘤微环境中功能复杂且来源多样的核心基质细胞[6]。它们并非一个均一的群体,而是具有显著的异质性。肝脏中的 CAF 主要来源于静息的肝星状细胞 (hepatic stellate cell,HSC) 的活化。在肝损伤或肿瘤信号如转化生长因子- β (transforming growth factor-β,TGF-β)、血小板衍生生长因子 (platelet-derived growth factor,PDGF) 刺激下,HSC 被激活并转化为肌成纤维细胞样表型的CAF[7]。此外,肝窦内皮细胞、门静脉成纤维细胞甚 至 上 皮 细 胞 通 过 上 皮 - 间 质 转 化 (epithelialmesenchymal transition,EMT)也可能转变成CAF。根据细胞表面标志物[如 α-平滑肌肌动蛋白 (α-smooth muscle actin, α -SMA)、 成 纤 维 细 胞 活 化 蛋 白(fibroblast activation protein,FAP)、血小板衍生生长因子受体 β (platelet-derived growth factor receptor β,PDGFRβ)]和功能差异,CAF 可分为不同的亚群,例如具有强促肿瘤功能的肌成纤维细胞样 CAF(myofibroblast-like CAF,myCAF) 和具有免疫调节功能的炎症性 CAF (in-flammatory CAF,iCAF),它们对 LCSC 的调控作用可能各不相同[8]。值得注意的是,近期基于单细胞与空间转录组的研究进一步细化了 CAF 的分类,并揭示 iCAF 在肿瘤核心与边缘的富集程度不同,其与 LCSC 的空间邻近性更强,提示 iCAF 可能在生态位中扮演更直接的支持角色[9]。
CAF 能够通过多种机制维持和增强 LCSC 的干性。首先,CAF 分泌大量的生长因子和细胞因子,如 肝 细 胞 生 长 因 子 (hepatocyte growth factor,HGF)、TGF-β 等。其中 HGF 能通过与其受体 cMet 结合,激活下游的磷脂酰肌醇 3-激酶 (phosphatidylinositol 3-kinase, PI3K)/蛋 白 激 酶 B(protein kinase B,AKT) 和丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)通路,直接促进 LCSC 的自我更新[10]。而 TGF-β 信号不仅能诱导 EMT,赋予细胞干细胞样特性,还能与 Wnt/β连环蛋白 (β-catenin) 等信号通路发生交叉对话,协同增强 LCSC 的干性[11]。其次,CAF 是 ECM 成分的主要生产者。重构的 ECM 不仅为 LCSC 提供了物理支架,其通过整合素等黏附分子传递的机械信号和生化信号,对干性维持也至关重要。例如,纤 连 蛋 白 可 通 过 整 合 素 α5β1 激 活 黏 着 斑 激 酶(focal adhesion kinase,FAK)/Src 激酶(Src kinase,Src) 信号通路,促进 LCSC 的富集[12]。此外,CAF还能通过其活跃的代谢活动,分泌乳酸、酮体等代谢物,创造出一个酸性的、营养重塑的微环境,这种代谢应激也已被证实可筛选具有更强可塑性和耐药性的 LCSC 亚群[13]。
1.2 免疫细胞在肝癌干细胞生态位中的作用
肿瘤微环境中的免疫细胞扮演着复杂的双重角色。在理论上,它们本应是识别并清除肿瘤细胞的主力军。然而,在成熟的肝癌组织中,LCSC 生态位通过一系列精密的免疫抑制机制,成功重塑了浸润的免疫细胞,使其从攻击者转变为维护生态位稳定、促进 LCSC 存活的支持者[14]。以下将聚焦于生态位中两个最具代表性的免疫抑制群体——TAM 与Treg,探讨它们如何被 LCSC 及其生态位招募、极化与利用,进而通过直接或间接的机制,为 LCSC构筑一个坚实的免疫抑制屏障。
1.2.1 肿瘤相关巨噬细胞的极化及其对肝癌干细胞的影响 TAM 是肿瘤微环境中数量最丰富、功能最关键的免疫细胞群之一[15]。它们主要来源于外周血中的单核细胞,在肿瘤分泌的趋化因子[如 C-C基序趋化因子配体 2(C-C motif chemokine ligand 2,CCL2)]作用下被招募至肿瘤组织,并在局部细胞因子的影响下极化为不同的功能亚型。它们不仅是清除癌细胞和呈递抗原的重要效应细胞,更在肿瘤的血管生成、免疫抑制、组织重塑以及维持 CSC 干性等多个环节中扮演着复杂而关键的角色[16]。通常,TAM 表现出类似于 M2 巨噬细胞的表型,M2 型TAM 能够通过分泌一系列因子直接或间接支持LCSC。例如,TAM 分泌的乳脂球状 EGF 因子 8(milk fat globule-EGF factor 8,MFG-E8) 可通过激活信号转导与转录激活因子 3 (signal transducer and activator of transcription 3,STAT3) 和 Hedgehog 信号通路,正反馈调节 LCSC 的自我更新[17]。此外,TAM 还高表达程序性死亡配体 1 (programmed cell death ligand 1,PD-L1),通过与肿瘤浸润 CD8+ T 细胞表面的程序性死亡蛋白 1 (programmed cell death protein 1,PD-1) 结合,直接传递抑制性信号,导致 T 细胞凋亡与功能耗竭,间接为 LCSC 创造免疫抑制微环境[18]。最新研究揭示,肝癌中的 TAM 是一个包含多个转录状态亚群的连续谱系,其中一群高表达分泌型磷蛋白 1 (secreted phosphoprotein 1,SPP1) 和载脂蛋白 E (apolipoprotein E,APOE) 的TAM 亚群与 LCSC 在空间上紧密相邻,其基因特征与最差的临床预后显著相关[19]。这一发现直接将特定的 TAM 亚群与干性维持功能联系起来。
1.2.2 调节性 T 细胞与肝癌干细胞的相互关系 Treg是一类具有免疫抑制功能的 CD4+T 细胞亚群。LCSC 能够通过高表达并分泌趋化因子 CCL22 及CCL5,与 Treg 表面 CCR4 形成趋化轴,主动招募Treg 至其周围,构建局部免疫抑制生态位;阻断CCL22-CCR4 信号可在体内外显著降低 Treg 浸润并恢复抗肿瘤免疫[20]。聚集的 Treg 还能通过分泌抑制性 细 胞 因 子 白 细 胞 介 素 (interleukin, IL) -10、TGF-β 和腺苷,以及通过细胞接触依赖性机制[如细胞 毒 性 T 淋 巴 细 胞 相 关 抗 原 4(cytotoxic Tlymphocyte-associated antigen 4,CTLA-4)与 CD80/CD86 结合],强烈抑制效应 T 细胞和自然杀伤(natural killer,NK) 细胞的抗肿瘤活性,从而为LCSC 营造一个强大的免疫抑制屏障。此外,还有研究表明肿瘤内 Treg 的数量与 LCSC 标志物 (如CD133、CD44) 的表达水平呈正相关,且预示着较差的预后[21]。Treg 的功能维持同样高度依赖生态位的代谢支持。最新研究表明,肿瘤微环境中丰富的乳酸和脂质代谢产物,能够通过增强 Treg 的氧化磷酸化和脂肪酸氧化 (fatty acid oxidation,FAO) 代谢程序,为其在营养竞争和压力环境下存活并发挥强大抑制功能提供能量基础[22]。
1.3 内皮细胞与肝癌干细胞生态位的关系
血管系统不仅是肿瘤生长所需的氧气和营养物质的核心输送通道,其内壁的内皮细胞本身也是构成功能性生态位的关键基质细胞。在肿瘤中,内皮细胞通过分泌特定的信号分子和提供直接的细胞间接触,主动调控着肿瘤细胞的命运。同时,肿瘤血管通常结构异常、功能不全,这种独特的血管形态直接塑造了肿瘤微环境中缺氧、高压的物理化学特征[23]。因此,理解内皮细胞的功能及其所构成的血管网络特性,是解析 LCSC 生态位空间定位和功能维持的基础。例如,在胶质瘤模型中,内皮细胞释放 的 NO 经 环 磷 酸 鸟 苷 (cyclic guanosine monophosphate,cGMP)/蛋白激酶 G (protein kinase G,PKG)激活 Notch 信号,显著提升 CSC 的自我更新与成瘤能力[24]。不成熟且高通透性的肿瘤血管导致组织间隙高压和缺氧,缺氧微环境通过稳定缺氧诱导因子-1α(hypoxia-inducible factor-1α,HIF-1α)转录因子,直接上调性别决定区 Y 框蛋白 2 (sexdetermining region Y-box 2,SOX2)、Nanog 同源盒(Nanog homeobox,NANOG)、八聚体结合转录因子4 (octamer-binding transcription factor 4,OCT4) 等干性基因,并诱导细胞周期阻滞,使 LCSC 维持于静息状态,从而逃避周期特异性化疗药物的杀伤[25]。此外,LCSC 与内皮细胞之间还存在密切的双向互作:LCSC 通过分泌血管内皮生长因子 (vascular endothelial growth factor,VEGF)、血管生成素 2(angiopoietin 2,ANGPT2) 等促血管因子诱导内皮增殖与血管生成;反之,活化的内皮细胞上调 E-选择素、血管细胞黏附分子 1 (vascular cell adhesion molecule 1,VCAM-1),与 LCSC 表面唾液酸路易斯 X 抗原 (sialyl Lewis X,sLeX)、迟现抗原 4(very late antigen-4,VLA-4) 等配体结合,介导其在内皮上的黏附-滞留-跨内皮迁移,从而促进肿瘤扩散与转移定植[26]。这种双向交流构成了一个正反馈循环,共同维持着生态位的稳定和功能。此外,近期有研究聚焦于转移前生态位的形成,发现原发性肿瘤中 LCSC 释放的外泌体可远程调控远处器官(如肺) 的内皮细胞,使其上调黏附分子和促炎因子,为循环 LCSC 的捕获和定植做好准备[27]。
综上所述,CAF、免疫细胞 (TAM,Treg) 和内皮细胞在 LCSC 生态位中扮演着不同但互补的角色,它们共同构成了一个功能协同的生态系统。更为关键的是,这些细胞之间存在着密集的正反馈循环。例如,CAF 分泌的因子可促进 TAM 的 M2 极化及 Treg 的招募,而 TAM 又可反馈促进 CAF 的活化与血管生成[28];内皮细胞形成的异常血管则通过造成缺氧,同时激活 CAF 并诱导 LCSC 干性[24]。这种网络化的互作,而非单一细胞的作用,使得生态位具备了强大的稳健性和促肿瘤功能。因此,未来针对生态位的治疗策略,必须从靶向单一细胞类型转向破坏整个协同网络,方能取得突破。
前述各类生态位细胞对 LCSC 的精密调控,依赖于一个高度复杂的分子信号网络来实现。该分子网络的核心组成部分,主要包括多条在进化上高度保守的信号通路,以及发挥着精细调控作用的非编码 RNA。以下将对其中最为关键的几条信号通路进行详细阐述。
2.1 经典信号通路在肝癌干细胞生态位中的作用
在 LCSC 生态位中,多条在进化上保守的信号通路被异常激活,它们构成了调控 LCSC 干性行为的 分 子 主 干 。 其 中 , Wnt/β -catenin、 Notch 和Hedgehog 信号通路的作用尤为突出。
2.1.1 Wnt/β-catenin 信号通路的核心地位 Wnt/βcatenin 通路是一条在进化上高度保守的信号通路,在胚胎发育、组织稳态及干细胞自我更新中发挥基础性作用。在 LCSC 中,该通路保持高活性,维持其自我更新、致瘤性及耐药表型[29]。在生态位中,CAF 及内皮细胞分泌的 Wnt3a,会和 LCSC 表面的卷曲蛋白 (Frizzled,Fzd) 受体及低密度脂蛋白受体 相 关 蛋 白 5/6 (low-density lipoprotein receptorrelated protein 5/6,LRP5/6) 共受体结合,抑制 βcatenin 降解复合体,促使 β-catenin 稳定积累并入核;核内 β -catenin 与 T 细胞因子 (T cell factor,TCF)/淋 巴 增 强 因 子 (lymphoid enhancer factor,LEF) 转录因子结合,启动 c-Myc、细胞周期蛋白D1 (cyclin D1, CCND1)、 分 化 簇 44 (cluster of differentiation 44, CD44)、 上 皮 细 胞 黏 附 分 子(epithelial cell adhesion molecule,EpCAM) 等靶基因转录,共同调控 LCSC 的自我更新、增殖与代谢重编程[30]。且生态位中的炎症信号 (如 IL-6) 和缺氧信号 (HIF-1α) 可与 Wnt 通路形成正反馈,进一步放大其效应。
2.1.2 Notch 信号通路的协同作用 Notch 信号通路通过受体与相邻细胞膜上的配体相结合来传递信号,从而直接调控细胞的增殖、分化和凋亡[31]。Notch 通路参与 LCSC 生态位的调控以维持 LCSC的干性。研究表明,邻近的内皮细胞或活化的 CAF其表面高表达锯齿状配体 1 (Jagged canonical Notch ligand 1, JAG1) 或 Delta 样 配 体 4 (Delta-like ligand 4,DLL4) 等配体,它们与 LCSC 表面的Notch 受体结合,有效激活了 LCSC 内的 Notch 信号 。 被 激 活 的 Notch 胞 内 结 构 域 (Notch intracellular domain,NICD) 通过上调 HES 家族碱性螺旋-环-螺旋转录因子 (HES family bHLH transcription factor,HES)/HEY家族碱性螺旋-环-螺旋转 录 因 子 (HEY family bHLH transcription factor,HEY)等靶基因,有力地抑制了 LCSC 的分化进程,使其维持于未分化的干细胞状态,并显著增强了其对化疗药物的耐药性[32]。尤为重要的是,Notch 通路并非独立发挥作用,它与 Wnt/β-catenin 等其他关键干性通路存在着密集的交叉串扰。例如,研究发现 CAF 高表达的 JAG1 与 LCSC 的富集及索拉非尼耐药密切相关。在机制上,Wnt/β-catenin 通路的关键转录复合物 β-catenin/转录因子 4 (transcription factor 4,TF4) 可直接上调 Jagged1 的转录;反之,Notch 的效应分子 NICD 也能反馈调节 β-catenin 的活性[33]。这种双向互作的网络化调控,使得 Notch与 Wnt 通路能够协同作用,共同强化 LCSC 的自我更新、EMT 进程及耐药表型。因此,联合靶向Notch 与 Wnt/β-catenin 信号已成为克服肝癌治疗耐药的一个极具前景的新策略。
2.1.3 Hedgehog 信号通路的关键作用 Hedgehog信号通路是一条在胚胎发育、组织稳态及干细胞干性调控中至关重要的进化上高度保守的信号通路[34]。在 LCSC 生态位中,Hedgehog 通路通过旁分泌机制被激活。生态位中的间质细胞 (如 CAF) 可分泌Hedgehog 配体,这些配体作用于 LCSC 表面的patched 同源物 1 (patched 1,PTCH1) 受体,通过上述机制解除对平滑蛋白 (smoothened,SMO) 的抑制,最终导致 GLI 家族锌指蛋白 (GLI familyzinc finger,GLI) 转录因子的激活[35]。Hedgehog 通路的激活不仅直接促进 LCSC 的自我更新,还能够通过间接方式影响生态位。例如,Hedgehog 信号能够驱动 CAF 分泌更多的 TGF-β 并促进细胞外基质(如胶原蛋白)的沉积,从而从信号和物理结构两方面进一步固化并强化这一促干性微环境[36]。尽管针对 SMO 的抑制剂在基底细胞癌等疾病中已获批应用,但在肝癌的临床试验中疗效有限,因为仅部分肝癌患者存在经典的配体-SMO-GLI 轴激活;更多情况下,Hedgehog 通路的激活表现为下游转录因子GLI 的非经典激活,即由其他通路[如 TGF-β 信号通路、Kirsten 大鼠肉瘤病毒癌基因同源物 (Kirsten rat sarcoma viral oncogene homolog,KRAS) 信号通路]或细胞应激信号直接驱动,完全独立于上游SMO[37],这就提示靶向下游 GLI 转录因子本身可能是更具潜力的方向。
2.2 非编码 RNA 对肝癌干细胞生态位的调控
除了上述由蛋白质构成的经典信号通路,近年来,非编码 RNA 作为一种重要的表观遗传调控分子,被发现在精细调控 LCSC 生态位中发挥着不可忽 视 的 作 用 。 其 中 , 微 小 RNA (microRNA,miRNA) 和 长 链 非 编 码 RNA (long non-codingRNA,lncRNA)是目前研究最为广泛的两类,它们在肿瘤发生发展与干细胞调控中尤为重要。
在 LCSC 生态位中,特定的非编码 RNA 表达谱发挥着关键的调控作用。例如,miR-200 家族成员 (如 miR-200a、miR-200c) 通过直接靶向转录因子 ZEB1 和 ZEB2,抑制 EMT 过程,其低表达通常与 LCSC 特性的获得和不良预后相关[38]。另一方面,一些非编码 RNA 则扮演着促干性角色。例如,miR-181 家族在 LCSC 中高表达,通过直接靶向抑癌基因 NLK 和 RASSF1,解除对 Wnt/β-catenin 信号的抑制,从而增强 LCSC 的自我更新能力并诱导索拉非尼耐药[39]。
综上所述,LCSC 生态位的分子调控网络是一个由经典信号通路与非编码 RNA 构成的高度协同的动态系统。分子调控网络之间互相串扰,共同赋予了 LCSC 生态位强大的稳健性和可塑性,使其能够有效应对治疗压力。
传统“一刀切”式的抗癌策略往往难以根除受到生态位庇护的 LCSC。针对生态位本身进行治疗干预,旨在破坏 LCSC 赖以生存的庇护环境,使其对常规治疗敏感,或直接消除其生存依赖,已成为具有前景的治疗新范式[40]。基于前述对生态位结构与功能的理解,当前治疗药物的干预策略主要分为两个方面:其一是靶向生态位的组成细胞,通过耗竭、重编程或从功能上抑制特定的基质细胞,来瓦解生态位的结构基础;其二是靶向其分子调控网络,通过抑制关键信号通路或利用非编码 RNA 等手段,干扰维持 LCSC 干性的核心信号。此外,本文还介绍了一种诱导 LCSC 向功能成熟、增殖能力有限的肝细胞分化的诱导分化治疗策略。
3.1 靶向生态位的组成细胞
鉴于 CAF、免疫细胞 (TAM、Treg) 及内皮细胞在生态位中的核心作用,它们自然成为了干预策略的首要靶点。针对这些细胞的治疗,旨在通过耗竭、功能重编程或阻断其与 LCSC 的通讯,来瓦解生态位的结构与功能支柱,从而将 LCSC 暴露于治疗攻击之下。
3.1.1 针对肿瘤相关成纤维细胞的干预方法 基于前述 CAF 在构建 LCSC 生态位中发挥的作用,靶向CAF 的策略主要分为三类。1) 抑制 CAF 活化:旨在从源头上减少促肿瘤 CAF 的丰度,使用 TGF-β受体抑制剂 (如 galunisertib) 或血管紧张素Ⅱ受体阻滞剂 (如氯沙坦) 可抑制 HSC 向 CAF 的分化,减少促肿瘤 CAF 的数量[41]。然而,galunisertib 在胰腺癌等实体瘤的临床试验中未达到主要终点,这一结果表明,在复杂的肿瘤微环境中,针对单一信号通路的干预可能不足以改变已建立的生态位稳态,未来可能需要联合靶向 CAF 活化的多条上游通路[42]。例如基于 CAF 特异性标志物 FAP 的靶向治疗,利用镓-68-标记成纤维细胞活化蛋白抑制剂(Gallium-68-labeled fibroblast activation protein inhibitor,68Ga-FAPI)进行 PET/CT 显像可无创、高特异性地可视化 CAF 丰度与分布[43];在此基础上,使用治疗性核素 (如 177Lu) 标记的 FAPI 配体进行内照射治疗,这种方法已在早期临床试验中显示出对多种实体瘤(包括肝癌)的治疗潜力[44]。2)靶向CAF 分泌因子:针对 CAF 分泌的 HGF 或 PDGF 的中和抗体或小分子抑制剂 (如可抑制 c-Met 的crizotinib,可阻断 PDGFR 的 imatinib),可阻断这些 信 号 对 LCSC 的 促 进 作 用[45]。 3) 破 坏 CAFLCSC 相互作用:靶向 CAF 产生的 ECM 成分 (如针对纤连蛋白的抗体) 或 LCSC 表面的整合素 (如针对整合素 α5β1 的抑制剂 ATN-161),可以破坏二者间的黏附信号,使 LCSC 失去锚定支持[46]。这些策略在临床前模型中均已显示出抑制肿瘤生长和清除 LCSC 的潜力。
3.1.2 调节免疫细胞功能的治疗手段 LCSC 生态位中的免疫抑制状态是维持 LCSC 功能的关键,因此,重塑免疫微环境成为核心治疗策略。当前的研究方向主要集中在干预关键的免疫抑制细胞群体及其相互作用上。1) 重编程 TAM:集落刺激因子 1受体 (colony-stimulating factor 1 receptor,CSF1R)抑制剂(如 PLX3397、BLZ945)可有效消耗 TAM。此外,CD40 激动性抗体、Toll 样受体激动剂等可将TAM 从 M2 型极化为具有抗肿瘤活性的 M1 型[47]。2) 靶 向 Treg: 其 中 抗 CCL5/CCR5 药 物 (如maraviroc) 可抑制 Treg 的募集。抗 CTLA-4 抗体(如伊匹木单抗)可通过抗体依赖性细胞介导的细胞毒性作用清除肿瘤内的 Treg,解除免疫抑制[48]。另外有研究发现,肿瘤内 Treg 高度依赖 FAO 供能,而 肉 碱 棕 榈 酰 转 移 酶 1A (carnitine palmitoyltransferase 1A,CPT1A) 是 FAO 限 速 酶 ; 使 用CPT1A 抑制剂 etomoxir 可选择性地削弱肿瘤内 Treg的功能并降低其存活率,而不影响外周免疫稳态,这为选择性免疫调节提供了极具前景的新靶点[49]。3) 基 于 免 疫 检 查 点 抑 制 剂 (immune checkpoint inhibitor,ICI) 的联合治疗:虽然 LCSC 和生态位免疫细胞高表达 PD-L1,但单用 PD-1/PD-L1 抑制剂在肝癌中的响应率有限。将其与靶向 TAM、Treg或直接靶向 LCSC 的药物联用,有望协同激活抗肿瘤免疫并清除 LCSC。例如,抗 PD-1 抗体联合CSF1R 抑制剂或 Wnt 通路抑制剂,在临床前模型中显示出协同抗肿瘤效果[50]。
3.1.3 基于抗血管生成治疗的生态位干预策略 如前文所述,内皮细胞不仅是营养输送的管道,更是构成 LCSC 生态位、通过血管分泌信号直接支持干性的活跃组分。因此,以 VEGF 抑制剂 (如贝伐珠单抗) 和多靶点酪氨酸激酶抑制剂 (tyrosine kinase inhibitor,TKI)(如仑伐替尼) 为代表的抗血管生成药物,其作用远不止于抑制肿瘤血管新生、阻断营养供应。它们通过靶向血管系统,直接攻击了由内皮细胞构成的这一生态位支柱,破坏其提供的生存信号。然而,这种治疗手段是一把双刃剑:在初期抑制血管的同时,也可能导致更严重的缺氧,从而诱导 HIF-1α 介导的 LCSC 富集和耐药,这解释了其疗效的局限性[51]。为了克服这一矛盾,将抗血管生成治疗与其他靶向策略联用已成为必然方向。其核心理念是从单纯的血管抑制转向血管正常化与生态位瓦解相结合。例如,临床已应用的仑伐替尼联合帕博利珠单抗的方案,其良好的疗效正源于仑伐替尼带来的血管正常化改善了肿瘤免疫微环境,与免疫检查点抑制剂协同作用,间接破坏了 LCSC 的免疫抑制生态位[52]。
3.2 靶向分子调控网络
相较于靶向特定的生态位组成细胞,直接干预其下游的分子调控网络,有望从根源上瓦解维持LCSC 干性的信号基础。该策略主要集中于两大方面:一是上述细胞相互作用所依赖的关键信号通路(如 Wnt、Notch、Hedgehog) 的抑制剂开发;二是针对调控这些通路的非编码 RNA 的靶向治疗。
3.2.1 针对信号通路的抑制剂开发 针对 Wnt/β-catenin、Notch 和 Hedgehog 这几条在 LCSC 生态位构建中发挥核心作用的信号通路,相应的抑制剂开发正持续取得进展。在靶向 Wnt/β-catenin 通路方面,由于该通路在正常组织稳态中的重要作用,直接抑制 β-catenin 面临挑战,当前策略主要聚焦于通路上游,例如通过 PORCN 抑制剂 (如 LGK974、ETC-159) 阻止 Wnt 配体的棕榈酰化及其分泌,这类药物在携带特定基因改变的临床前模型中已显示疗效;同时,针对肿瘤特异性共受体或下游转录复合物的探索也在进行中[53]。对于 Notch 通路,γ-分泌酶抑制剂 (如 MK-0752) 虽能广泛抑制 Notch 信号,但其胃肠道毒性限制了临床应用,推动研发转向更具选择性的 Notch 受体拮抗性抗体或 DLL4 特异性抑制剂[54]。在 Hedgehog 通路领域,已获批用于 基 底 细 胞 癌 的 SMO 抑 制 剂 (如 vismodegib、sonidegib) 在肝癌临床试验中效果不尽如人意,这可能与旁路激活或肿瘤异质性有关,因此靶向下游GLI 转录因子的直接抑制剂 (如 GANT61) 正成为新的研究方向[55]。总体而言,针对这些关键通路的抑制剂研发正朝着提升选择性、克服耐药性和降低毒性的方向不断深化。
3.2.2 非 编 码 RNA 作 为 治 疗 靶 点 的 潜 力 以miRNA 和 lncRNA 为代表的非编码 RNA,因其在LCSC 生态位中发挥的关键调控作用,已成为一类新兴的治疗靶点。目前主要的干预策略包括通过递送合成的 miRNA 模拟物来恢复抑癌 miRNA (如miR-34a)的功能,或使用反义寡核苷酸等核酸抑制剂抑制促癌 miRNA (如 miR-21) 的活性。这些方法在临床前研究中已被证实能够有效抑制 LCSC 的自我更新、增强化疗敏感性,并改善肿瘤免疫微环境[56]。该类药物如 MRX34 (miR-34a 模拟物) 在早期临床试验中因免疫相关不良事件而终止,但其为基于 miRNA 的治疗路径提供了重要的概念验证。当前的研究重点正转向开发更安全、高效的靶向递送系统(如基于纳米颗粒的载体),以提升治疗的精准 度 和 耐 受 性[57]。 同 时 , 对 lncRNA (如 H19、MALAT1)作为潜在治疗靶点的探索也在不断深入,这些分子在调控 LCSC 干性及生态位稳态中同样发挥着重要作用,有望为未来治疗策略提供新的突破口[58]。
3.3 诱导肝癌干细胞分化
与传统旨在根除肿瘤细胞的策略不同,诱导分化治疗代表了一种重塑肿瘤细胞命运的新范式。诱导分化治疗可以促使 LCSC 向成熟的肝细胞分化,并永久性丧失其自我更新、致瘤及转移的干性潜能[59]。该策略有望避免传统疗法对 LCSC 的负向选择压力,为克服治疗抵抗与复发提供了全新思路。
多种小分子化合物、细胞因子及基因调控手段已被证实在体外和体内模型中能有效诱导 LCSC 分化 。 1) 全 反 式 维 A 酸 (all-trans retinoic acid,ATRA):ATRA 是维生素 A 的衍生物,通过结合并激活维甲酸受体 (retinoic acid receptor,RAR),调控下游大量与细胞分化、增殖和凋亡相关的基因表达。在肝癌中,ATRA 可通过上调肝细胞核因子 4α(hepatocyte nuclear factor 4α,HNF4α)、CCAAT/增强子结合蛋白 α (CCAAT/enhancer-binding protein α,C/EBPα) 等肝细胞关键转录因子,同时抑制干细胞标志物如 NANOG、OCT4 的表达,从而诱导LCSC 向肝细胞样细胞分化,并显著降低其成瘤能力[60]。2) 抑瘤素 M (oncostatin M,OSM):OSM是 IL-6 家族细胞因子,通过激活 Janus 激酶 (Janus kinase,JAK)/STAT3 信号通路,在肝脏发育和再生中促进肝细胞分化。在肝癌模型中,OSM 被证明能有效诱导 LCSC 分化,其机制可能与 STAT3 介导的特定基因程序激活有关,且该作用可能独立于其促炎功能[61]。3)基因调控策略:强制表达关键肝细胞分化主导因子,如 HNF4α,可直接重编程 LCSC,使其获得成熟肝细胞的转录组和表型,是证明分化治疗可行性的有力概念验证[62]。图 1 总结了 LCSC生态位的细胞组成、分子调控网络及相关治疗策略。
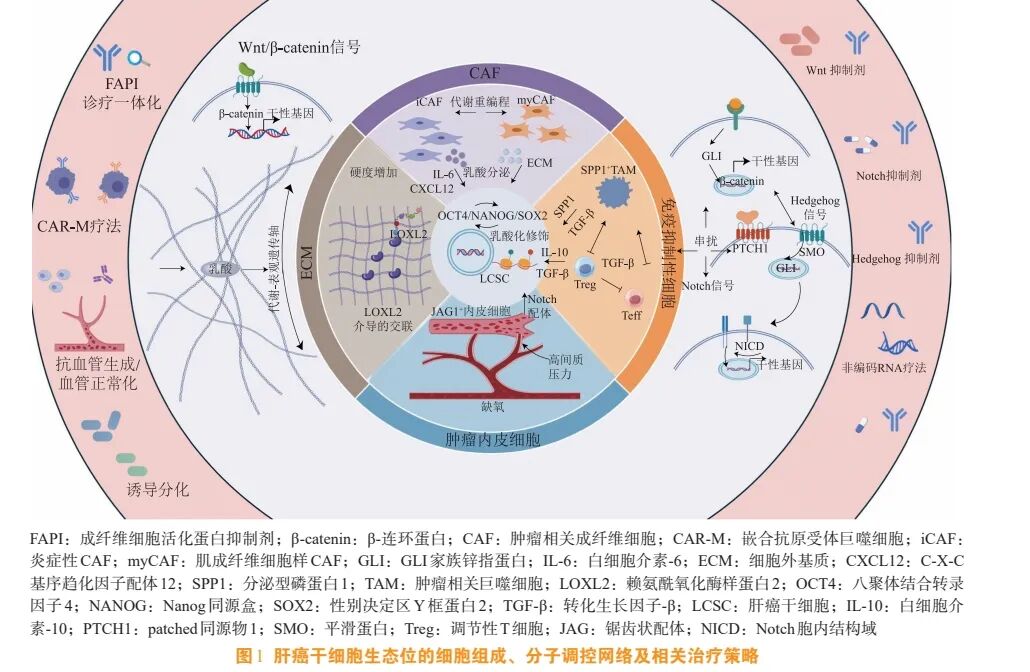
3.4 小结
总体而言,针对 LCSC 生态位的治疗策略已形成了清晰的脉络:其一是通过靶向 CAF、免疫细胞及内皮细胞等基质细胞组分以瓦解生态位的结构基础;其二是通过抑制关键信号通路及调控非编码RNA 以干预其分子调控网络;其三是通过诱导LCSC 分化以使其丧失干性潜能。然而,生态位本身的复杂性决定了单一靶向策略往往难以取得理想效果。因此,未来治疗模式的突破必然依赖于联合策略的深度优化。这不仅包括生态位不同靶向药物之间的联合,更涵盖生态位靶向治疗与传统化疗、放疗、免疫治疗之间的时序性与空间性协同,在破坏生态位稳态的同时根除 LCSC,最终实现从根本上改善肝癌患者预后的目标。
过去十年,我们在理解 LCSC 生态位方面取得了显著进展。1)细胞图谱逐渐清晰:通过单细胞测序等技术,初步绘制了肝癌微环境中不同细胞亚群的图谱,揭示了 CAF、TAM 等细胞的异质性及其与 LCSC 的潜在关联[63]。2) 关键通路得到确认:Wnt、 Notch、 Hedgehog 等 核 心 信 号 通 路 在 维 持LCSC 干性及生态位稳态中的作用被广泛证实。3)治疗策略不断涌现:大量临床前研究证明了靶向生态位各组分 (如 CAF、免疫细胞、信号通路) 及诱导分化策略在抑制肿瘤生长、克服耐药方面的潜力,为联合治疗提供了丰富的候选方案。
生态位的空间结构和细胞间相互作用的动态性是其核心特征,但目前的研究技术仍存在局限。1)空间分辨率:常规单细胞测序丢失了细胞的空间位置信息。发展单细胞空间转录组、多重免疫荧光成像、空间蛋白质组学等技术,对于解析生态位内细胞的确切邻近关系至关重要[64]。2) 体内实时监测:缺乏能够在活体动物中实时、动态观测 LCSC 与生态位细胞相互作用的技术。新型活体成像技术和报告基因系统的发展将有助于揭示生态位的动态演化过程。3) 体外模型:传统的 2D 细胞培养无法模拟生态位的复杂性。开发更先进的 3D 类器官共培养模型、器官芯片等,将提供更贴近生理状态的研究平台[65]。
近年来临床试验的反馈结果,修正了我们对LCSC 生态位理论的理解。首先,由于针对 TGF-β等泛 CAF 信号的疗法临床效果有限,促使基础研究重新审视其异质性。随后有研究揭示,iCAF 亚群在LCSC 生态位中可能比产生基质的 myCAF 发挥着更为关键的作用[9]。其次,经典通路抑制剂容易引起耐药的现象,使得研究者们转而关注代谢层面。有研究发现,生态位中堆积的乳酸、酮体等不仅是代谢废物,它们还能通过调控表观遗传[如抑制组蛋白去乙酰化酶 (histone deacetylase,HDAC)]直接维持 LCSC 干性[27]。最后,抗血管药物联合免疫疗法的成功(如仑伐替尼联合帕博利珠单抗),标志着这种联合策略可通过重构生态位、逆转免疫抑制,实现免疫微环境的重编程[52]。这些临床反馈表明,生态位是一个高度可塑和代偿的系统。未来治疗策略必须基于对其动态性和异质性的新认识,并且通过持续的基础研究与临床反馈来优化。
LCSC 生态位是一个高度复杂、动态且功能强大的微环境单元。本综述系统梳理了该生态位的核心组成细胞,包括 CAF、TAM、Treg 和内皮细胞等,解析了它们通过直接接触或旁分泌方式与LCSC 构成的双向通讯网络。此外,还介绍了 Wnt/β-catenin、Notch、Hedgehog 等关键信号通路以及非编码 RNA 在这一网络中的核心调控作用,揭示了生态位内在的分子串扰机制;并在对生态位理解的基础上,提出了针对 LCSC 生态位的 3 种肿瘤治疗策略:1) 靶向细胞组成以瓦解其结构基础;2) 干预分子网络以阻断核心信号;3)诱导 LCSC 分化以使其丧失干性。
然而,该领域仍面临诸多挑战。生态位的动态性和高度异质性要求未来的基础研究必须更紧密地结合临床试验结果。展望未来,首先,应从临床实际问题出发,指导基础研究设计。可以通过空间多组学技术,直接比较应答者与非应答者、复发与未复发患者其 LCSC 生态位在细胞组成、空间构象和分子网络上的动态差异,从而揭示驱动治疗抵抗的核心生物学过程。其次,以基础研究新发现为指引,设计并优化临床联合治疗策略。未来可考虑设计临床试验将靶向 CAF 乳酸代谢的抑制剂、阻断 TAM招募的 CSF1R 抑制剂以及 PD-1 抑制剂三者联用,从代谢、细胞丰度和免疫检查点 3 个层面来系统解构免疫抑制生态位。最后,利用临床试验结果修正现有的理论认知,形成“临床-基础-临床”的良性循环,推动产生更为有效的治疗策略。
本文引文格式:
郭颖,韩雪丹,郑禄枫.肝癌干细胞生态位的调控机制及干预路径研究进展[J].药学进展,2026,50(3):227-236.
信息来源:药学进展




免责声明
“汇聚南药”公众号所转载文章来源于其他公众号平台,主要目的在于分享行业相关知识,传递当前最新资讯。图片、文章版权均属于原作者所有,如有侵权,请在留言栏及时告知,我们会在24小时内删除相关信息。
本平台不对转载文章的观点负责,文章所包含内容的准确性、可靠性或完整性提供任何明示暗示的保证。
喜欢的点个“看一看”和"喜欢"吧

不然微信推送规则改变,有可能每天都会错过我们哦~





