 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库Cancer Res:刘明洋等团队发现胰腺癌化疗耐药新策略
胰腺癌仍然是最致命的恶性肿瘤之一,对大多数患者而言,以吉西他滨为基础的化疗仍是主要治疗手段,但几乎普遍会出现耐药。胰腺癌的一个显著特征是其致密的富含成纤维细胞的基质,其中异质性的癌症相关成纤维细胞(CAF)在调控肿瘤生物学行为及治疗反应中发挥重要作用。
2026年6月17日,中国医学科学院北京协和医学院刘明洋和俄克拉何马大学Min Li共同通讯在
Cancer Research
在线发表题为
Mitophagy-Competent Cancer-Associated Fibroblasts Fuel Chemoresistance by Rewiring Pyrimidine Metabolism in Pancreatic Cancer
的研究论文。
该研究阐明了一种基质–代谢机制,通过该机制耐药性CAF促进吉西他滨耐药。作者鉴定出一类具有线粒体自噬能力的CAF亚群,该亚群显著增强胰腺癌对吉西他滨的耐药性。
上皮–间质转化(EMT)转录因子ZEB1作为该CAF驱动耐药程序的主调控因子,在吉西他滨耐药CAF中上调,并通过SETD1A介导的H3K4甲基化发生表观遗传激活。ZEB1促进CAF中BNIP3依赖的线粒体自噬,导致核苷酸分泌增加。这些由CAF释放的核苷酸一方面竞争性抑制吉西他滨在肿瘤细胞中的掺入,另一方面为嘧啶代谢提供底物,从而增强嘧啶代谢通路。
与此同时,ZEB1在转录层面激活CXCL8,进而在肿瘤细胞中激活CXCR1/2–MEK/ERK信号通路,并通过RRM1/E2F1/G6PD轴进一步增强嘧啶代谢,从而整体降低吉西他滨的细胞毒性作用。值得注意的是,联合抑制CXCR1/2或G6PD与吉西他滨治疗可在体外和体内显著抑制肿瘤生长并恢复化疗敏感性。
综上,这些发现揭示了胰腺癌中一个关键的基质–代谢轴,将CAF的线粒体自噬活性与肿瘤细胞代谢重编程联系起来,并确定ZEB1及其下游网络为克服化疗耐药的可靶向关键节点。

胰腺癌预计将在2030年成为美国癌症相关死亡的第二大原因。尽管癌症检测与治疗不断进步,胰腺癌的5年生存率仍低于13%,主要原因包括其临床发现较晚、早期即发生转移以及对治疗具有显著抵抗性。
在胰腺癌的特征中,治疗耐药(无论是内源性还是获得性)在疾病进展和不良预后中起着关键作用。尽管靶向治疗和免疫治疗已改变了多种癌症的治疗格局,但其在胰腺癌中的获益仅限于少数患者,这些患者通常具有罕见的基因改变或特定的免疫表型。
对于绝大多数患者而言,吉西他滨(gemcitabine)为基础的化疗仍是系统治疗的核心。然而,在治疗过程中,几乎所有患者都会逐渐产生对吉西他滨的耐药性,从而严重限制其长期疗效。这种耐药性来源于肿瘤细胞内在机制以及肿瘤微环境(TME)介导的适应性反应共同作用的结果,这些机制共同降低药物敏感性并促进肿瘤存活。
胰腺癌肿瘤微环境的基因组与空间异质性导致其治疗反应存在差异。胰腺癌的TME以显著的促纤维化反应为特征,表现为大量细胞外基质沉积以及以癌症相关成纤维细胞(CAF)为核心的复杂基质成分。CAF不仅作为结构支架存在,还积极影响胰腺癌进展、免疫逃逸及治疗抵抗。
越来越多证据表明CAF在促进化疗耐药(尤其是吉西他滨耐药)中发挥关键作用。CAF通过多种机制介导耐药,包括代谢重编程以支持核苷酸生物合成、调控氧化应激反应,以及分泌旁分泌因子以保护肿瘤细胞免受化疗诱导损伤。
这些过程使癌细胞能够更高效地修复DNA损伤、逃避免细胞死亡并维持增殖,从而成为化疗效果受限的重要障碍。例如,CAF可通过释放含microRNA的细胞外囊泡抑制铁死亡,从而促进吉西他滨耐药;同时,CAF分泌的脱氧胞苷也可保护胰腺癌细胞免受吉西他滨的抗肿瘤作用。
然而,单纯广泛抑制CAF的策略在抗肿瘤效果上存在争议,因此理解CAF的异质性尤为重要。
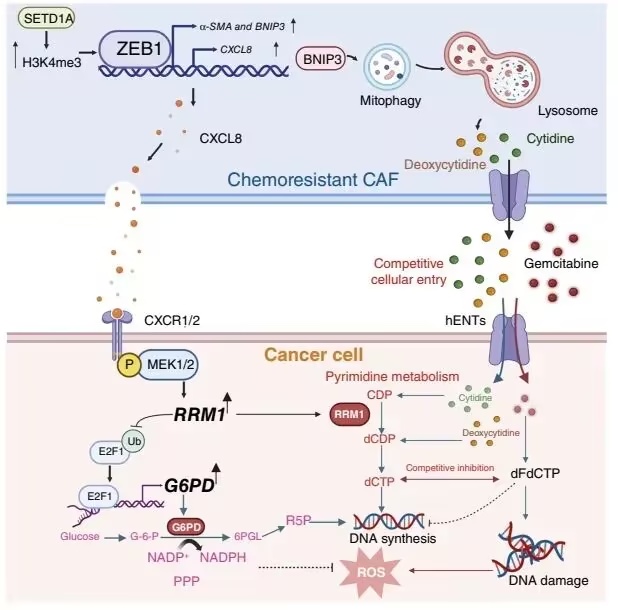
图1.全文总结图(摘自
Cancer Research
)
CAF是一个高度异质性的细胞群体,其来源包括常驻成纤维细胞、胰腺星状细胞、间充质干细胞等多种细胞类型。这种异质性形成了具有不同表型与功能的CAF亚群。部分CAF亚群具有抑癌作用,而另一些则促进肿瘤生长、转移及治疗耐药。
不同CAF亚群通过构建亚肿瘤微环境(subTME)和多细胞生态系统,共同塑造具有不同生物学特征的肿瘤环境。CAF的动态演化过程参与胰腺癌基质重塑。近年来单细胞转录组学研究逐渐揭示了具有特异分子特征和功能的CAF亚群。
胰腺癌中主要CAF类型包括肌成纤维样CAF(myoCAFs),其具有收缩和基质重塑能力并表达α-SMA、TAGLN等标志物;以及炎症型CAF(iCAFs),其以分泌功能为主并表达IL6、LIF等细胞因子。
此外,还发现了一些特殊CAF亚群,如促进神经浸润的神经侵袭相关CAF,以及具有血管拟态能力并通过旁分泌促进转移的内皮样CAF(endoCAF)。然而,哪些CAF亚群直接驱动化疗耐药,以及其具体信号网络仍未明确。解析这一复杂性对于开发能够选择性抑制促肿瘤CAF、同时保留甚至增强抑癌CAF功能的策略至关重要。
上皮–间质转化(EMT)转录因子ZEB1在癌症中已被广泛研究,其功能涉及肿瘤增殖、转移、耐药及免疫逃逸。然而,其在CAF中的作用仍知之甚少。目前仅有少数研究提示ZEB1可通过抑制炎症激活而促进结直肠癌中肌成纤维样特征。
ZEB1在肿瘤细胞与CAF中均可表达,但呈现异质性。在肿瘤细胞中,已有研究表明ZEB1可通过降低H3K27三甲基化并增加H3K4甲基化实现表观遗传激活,从而促进其转录表达。然而,CAF中ZEB1高表达的机制仍未见报道。
本研究中,作者从胰腺癌患者中分离原代CAF,并鉴定出一类通过自噬激活而促进化疗耐药的CAF亚群。作者发现ZEB1通过SETD1A依赖的H3K4甲基化上调表达,进而驱动CAF中的线粒体自噬(mitophagy)并增强CXCL8分泌。
机制上,ZEB1通过BNIP3介导的线粒体自噬促进核苷酸分泌,这些核苷酸一方面竞争性抑制吉西他滨(GEM)的细胞内掺入,另一方面为嘧啶代谢提供底物。同时,CXCL8通过激活CXCR1/2–MEK/ERK信号通路,并进一步通过RRM1–E2F1–G6PD轴促进肿瘤细胞嘧啶代谢,从而削弱吉西他滨的细胞毒性作用。
本研究揭示了一类在胰腺癌中具有不同功能影响的CAF群体,并阐明ZEB1在CAF中启动的全新机制:通过促进嘧啶代谢降低吉西他滨疗效,从而为克服胰腺癌化疗耐药提供潜在治疗靶点。
参考消息:https://doi.org/10.1158/0008-5472.CAN-25-5716





