五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库肝性脑病发病机制中的多器官交互作用

肝性脑病作为肝硬化的严重并发症,可显著缩短患者的中位生存期。其流行病学负担较重,30%~40%的肝硬化患者可进展为显性肝性脑病,50%~80%存在轻微型肝性脑病。近年来,随着肝硬化病因谱向代谢相关(非酒精性)脂肪性肝病及酒精性肝病转变,且常合并糖尿病与肌少症,肝性脑病的流行病学特征亦随之发生演变。
目前研究已表明肝性脑病是一种涉及多器官的全身性疾病,其主要发病机制(氨中毒、炎症、神经递质失衡以及氧化/硝化应激)虽奠定了理论基础,却难以完全解释肝性脑病的临床异质性及全身性病理生理变化。因此,本文以“多器官交互”为核心视角,逐一阐释在每种机制框架下,肠道、肝脏、肌肉、肾脏及大脑等器官如何构成动态的交互网络,深入探讨不同机制间的协同与交叉作用,并纳入新兴代谢风险因素,以期为肝性脑病的机制研究与多靶点治疗策略提供全新的整合性思路。
1氨中毒与多器官交互
氨中毒是肝性脑病发病机制的核心理论,该机制认为,肝性脑病患者的精神症状主要由肝功能障碍或门体分流引起的血氨水平升高所导致。其核心观点为:大量氨促使星形胶质细胞生成过量谷氨酰胺,引发脑细胞水肿,并通过抑制丙酮酸脱氢酶复合物等途径干扰大脑能量代谢,最终导致大脑功能失调。这一机制的直接证据来源于多数肝性脑病患者体内存在高氨血症,且降氨治疗具有一定的作用。尽管近年发现,血氨水平与临床症状并非完全线性相关,提示存在其他机制协同作用,但氨中毒至今仍是肝性脑病病理生理的核心机制。
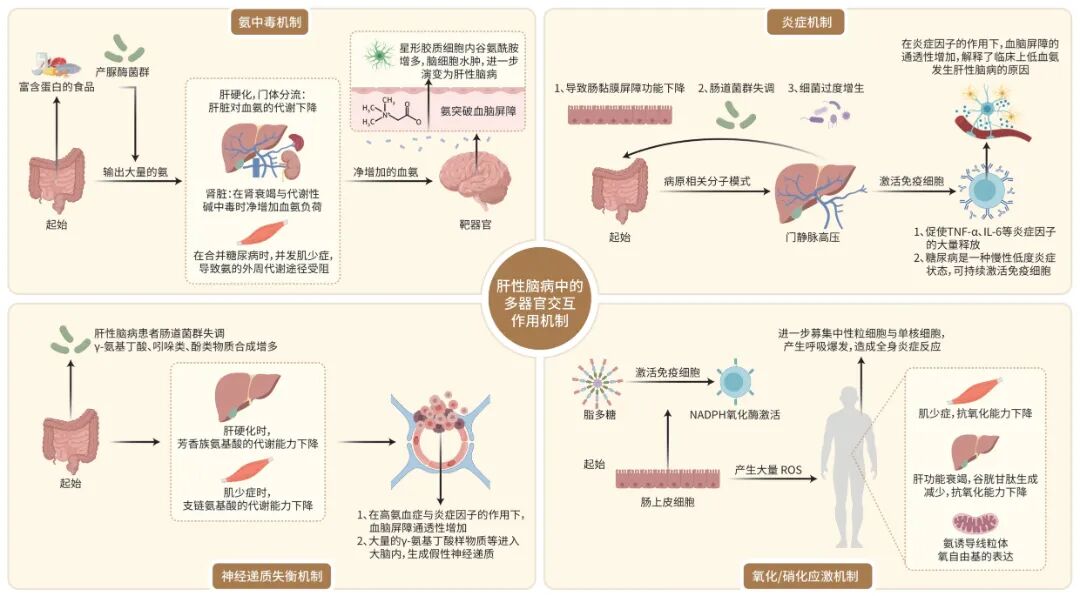
基于氨中毒理论,从多器官层面分析,氨代谢始于肠道。结肠内产脲酶菌群分解膳食蛋白质和尿素产生大量氨。肝性脑病患者普遍存在肠道菌群失调,具体表现为产氨菌过度增殖和微生物多样性降低,且这些变化与认知障碍严重程度相关。与此同时,肠道屏障功能受损促使氨易位入门静脉系统,从而大幅增加氨负荷。氨进入体循环后,肌肉通过谷氨酰胺合成途径固定血氨,是重要的外周解毒器官。然而,肝硬化晚期常并发肌少症,削弱了这一代偿途径。骨骼肌质量降低是预测肝性脑病发生风险的重要指标,因此通过营养支持和抗阻运动干预肌少症,可被视为降低血氨水平的治疗新思路。此外,糖尿病作为常见合并症,也是肌少症的重要诱因,进一步减弱骨骼肌的氨解毒能力。肾脏则通过近曲小管谷氨酰胺酶分解谷氨酰胺产氨,并在代谢性碱中毒时为维持氢离子储备而减少氨离子排泄,净增加血氨负荷,故其在病理状态下可加剧氨中毒。氨代谢的核心环节在肝脏,肝细胞尿素循环酶系完备,肝功能衰竭时尿素合成能力急剧下降,成为高氨血症的主要病因。氨随血入脑后主要在星形胶质细胞内代谢,生成谷氨酰胺引发渗透性水肿,同时通过抑制丙酮酸脱氢酶复合物等途径严重干扰能量代谢,共同导致神经网络功能失调,表现为认知障碍与意识状态改变(图1)。
注: TNF-α,肿瘤坏死因子α;IL-6,白细胞介素6;ROS,活性氧;NADPH,还原型烟酰胺腺嘌呤二核苷酸磷酸。
图1 肝性脑病中的多器官交互作用机制
传统氨中毒机制难以解释部分患者血氨水平与神经精神症状的分离现象。这提示高氨血症虽是肝性脑病的核心驱动因素,但其神经异常的表现依赖于多器官交互的病理背景。该临床异质性主要源于氨与其他因子(特别是系统性炎症)的协同作用,例如炎症可显著降低大脑对氨的耐受阈值并破坏血脑屏障,使患者在相对较低血氨水平下出现严重症状。未来研究需评估各器官对全身血氨的影响,并深入揭示氨与炎症介质、氧化应激在分子层面的交叉作用机制。
2炎症与多器官交互
炎症介质的作用是肝性脑病发病机制中的重要理论之一,强调系统性炎症和神经炎症在其发生发展中的核心作用。其核心观点为:肠道屏障功能障碍及菌群失调导致病原相关分子模式(PAMP)易位,激活免疫细胞并释放肿瘤坏死因子α(TNF-α)、白细胞介素(IL)1β等促炎细胞因子,引发全身性炎症反应。这些炎症介质可破坏血脑屏障,并与高氨血症产生协同效应,显著降低大脑对氨的耐受阈值,从而诱发或加重神经精神症状。
基于炎症发病机制,从多器官层面探讨,肠道是炎症反应的主要起源部位。肝硬化门静脉高压导致肠道黏膜屏障受损、菌群失调及细菌过度生长,形成“肠漏”,使大量PAMP易位至门静脉循环。当肝功能严重衰竭或存在广泛门体分流时,肝脏对PAMP的清除能力下降,导致其进入体循环。PAMP通过与免疫细胞模式识别受体结合,激活核转录因子κB等信号通路,促进TNF-α、IL-1β、IL-6等促炎细胞因子释放,形成持续性系统性炎症状态。此炎症状态不仅直接损害器官功能,还可下调脑血管内皮细胞紧密连接蛋白表达,显著增加血脑屏障的通透性,使血氨等神经毒素更易入脑,加剧神经损伤。值得关注的是,系统性炎症与高氨血症之间存在“协同毒性”效应,炎症可显著降低大脑对氨的耐受阈值,使患者在较低血氨水平下出现严重的神经精神症状,可能是低血氨患者发生肝性脑病的原因之一。临床观察可见,肝硬化患者常因感染等炎症状态出现神经心理恶化。糖尿病作为一种慢性低度炎症状态,可与肝硬化的固有炎症背景产生协同放大效应。胰岛素抵抗与高血糖可持续激活免疫细胞,促进促炎因子释放。当肝硬化合并糖尿病时,叠加的炎症状态进一步降低血脑屏障对神经毒素的耐受阈值,更易触发肝性脑病。因此,对于肝硬化患者积极控制血糖,不仅是管理代谢紊乱,更是预防肝性脑病发生的重要临床策略。最终,透过血脑屏障的炎症介质进入脑实质,激活小胶质细胞和星形胶质细胞(图1)。活化的小胶质细胞进一步释放TNF-α、IL-1β等细胞因子,形成自我延续、逐级放大的神经炎症环路。这种慢性神经炎症不仅直接导致神经元损伤与突触功能障碍,还可能通过破坏血脑屏障、影响脑血流调节等,共同扰乱神经网络活动。
炎症机制阐明了“肠-肝-脑轴”在肝性脑病中的核心地位,以及炎症反应与氨的协同毒性。然而,目前对于炎症信号在器官间传递的具体载体(如外泌体)及其特异性受体仍不明确。探索神经炎症启动与维持的精确细胞机制,以及如何与保护性炎症反应区分,将是开发精准抗炎策略的关键。
3神经递质失衡与多器官交互
神经递质失衡是从神经抑制角度阐释肝性脑病的发生机制,认为其大脑功能异常源于中枢神经系统内兴奋性与抑制性神经递质平衡的破坏,涉及假性神经递质取代、抑制性信号过度增强等过程。核心内容包括γ-氨基丁酸能抑制增强、谷氨酸兴奋性毒性以及芳香族氨基酸/支链氨基酸比例失衡导致的假性神经递质生成。具体而言,在肝功能严重受损时,肠道吸收的芳香族氨基酸(如苯丙氨酸、酪氨酸)在血液中浓度显著升高,并大量进入中枢神经系统。这些氨基酸在脑内经脱羧酶作用发生异常代谢:苯丙氨酸可转化为苯乙胺,并进一步生成苯乙醇胺,酪氨酸则可能被错误地羟化和脱羧生成章鱼胺。这些物质在化学结构上与正常的兴奋性神经递质相似,可竞争性地占据其受体位点,但其生物学活性远低于正常递质,从而严重干扰正常的儿茶酚胺能神经传递,导致神经抑制和意识障碍。随着研究深入,该机制又将视角拓展到肠道菌群衍生的多种神经活性物质,肠道内的这些神经活性物质可不依赖氨途径直接驱动神经炎症与认知损伤。
基于神经递质失衡这一发病机制与多器官层面分析,神经递质失衡起源于肠道。肝性脑病患者存在肠道菌群失调,产生γ-氨基丁酸及苯二氮䓬样物质的细菌过度增殖,加之屏障受损,使得这些神经活性物质易位进入门静脉循环。此外,肠道细菌代谢产生的酚类、吲哚类等物质也可入血干扰中枢神经系统功能。肌肉作为外周氨基酸代谢的关键场所,是支链氨基酸分解的主要部位,有助于降低芳香族氨基酸/支链氨基酸比值。肝硬化相关肌少症导致肌肉支链氨基酸代谢能力减弱,加剧比例失衡,促进假性神经递质生成;研究显示肌肉质量与肝性脑病严重程度呈负相关,其在维持氨基酸平衡中起重要作用。肝脏作为物质代谢的核心器官,其功能衰竭是神经递质失衡的始动和放大环节:首先,肝衰竭直接导致两大代谢缺陷,一是对芳香族氨基酸的分解能力下降,致使其在血中浓度升高;二是对γ-氨基丁酸等神经活性物质的清除能力急剧减弱。其次,肝功能不全所致的高氨血症,本身也会广泛干扰神经递质的合成与代谢。这些由肝脏病变引发的氨基酸失衡、神经活性物质蓄积及高氨血症等外周代谢紊乱,共同破坏了中枢神经系统的稳态。上述物质经过通透性增加的血脑屏障或者通过血液循环,最终协同作用于大脑,成为诱发神经递质失衡的核心外周驱动因素。在高血氨与炎症因子作用下,血脑屏障通透性增加,允许更多神经活性物质入脑。脑内高氨环境损害星形胶质细胞功能,抑制谷氨酸摄取,导致谷氨酸堆积及N-甲基-D-天冬氨酸受体过度激活,引发兴奋性毒性;同时,入脑的芳香族氨基酸转化为假性神经递质,竞争性干扰正常儿茶酚胺能传递,而增多的γ-氨基丁酸则强化抑制性信号,共同导致神经网络抑制增强及功能障碍。若患者合并糖尿病,其所致的高血糖与血糖波动可影响脑能量代谢底物供应,且在糖尿病与肝性脑病并存时,血脑屏障通透性进一步增加,使更多γ-氨基丁酸样物质及芳香族氨基酸入脑,增强神经抑制并提供假性神经递质底物,共同导致神经网络功能失调(图1)。
该机制强调了神经活性物质具有全身性来源。但传统研究过于聚焦γ-氨基丁酸/谷氨酸系统。近年发现的肠道菌群衍生神经活性物质(如苯乙胺),被证实可不依赖氨途径直接驱动神经炎症和认知损伤,这对以氨和经典递质为主的传统理论构成了有力挑战。
4氧化/硝化应激与多器官交互
在氧化/硝化应激的环境中,大脑的细胞损伤源于活性氧(ROS)与活性氮(RNS)过度产生。该机制认为,氧化与抗氧化系统失衡导致ROS/RNS大量积累,进而引发线粒体功能障碍、DNA损伤等,最终导致星形胶质细胞损伤与神经元死亡。其核心观点为氧化/硝化应激是氨中毒和炎症介质诱导的执行机制。在肝性脑病患者中,星形胶质细胞内氧化应激标志物升高,并存在硝化应激标志物3-硝基酪氨酸的积聚,其水平与疾病严重程度相关。
基于氧化/硝化应激发病机制,从多器官层面探讨,全身多器官共同参与了肝性脑病中氧化与抗氧化失衡。肠道是启动全身氧化应激的关键部位,肝硬化门静脉高压导致肠道黏膜屏障受损,内毒素易位至门静脉循环。脂多糖(LPS)可激活肠上皮细胞及免疫细胞中的还原型烟酰胺腺嘌呤二核苷酸磷酸氧化酶,产生ROS,并进一步激发全身性炎症反应,募集中性粒细胞和单核细胞发生“呼吸爆发”,产生大量ROS与RNS。肌肉不仅是氨解毒的场所,也是抗氧化物质合成与储存的外周组织。发生肌少症时,肌肉的抗氧化能力显著减弱,无法有效清除循环活性分子,从而削弱其对全身氧化还原平衡的调节能力。肝脏是合成谷胱甘肽的主要器官,功能衰竭时谷胱甘肽合成急剧减少,机体清除ROS的能力随之下降。最终,外周器官的紊乱共同作用于大脑:氨诱导线粒体功能障碍,增加ROS的产生;并与炎症因子协同,诱导星形胶质细胞和小胶质细胞中诱导型一氧化氮合酶高表达,产生过量一氧化氮。一氧化氮与超氧阴离子反应生成强毒性的过氧亚硝酸盐,后者可硝化蛋白质酪氨酸残基形成3-硝基酪氨酸,抑制星形胶质细胞谷氨酸转运体的功能,破坏谷氨酸稳态并损害线粒体功能(图1)。这促使脑内ROS/RNS大量产生,诱导线粒体功能障碍、蛋白质硝化及神经炎症,共同导致星形胶质细胞损伤与神经功能损害。糖尿病相关的高血糖是加剧氧化/硝化应激的关键因素。高血糖状态可导致线粒体功能障碍和ROS过量生成,与氨诱发的氧化应激产生协同效应。值得注意的是,在糖尿病合并肝性脑病的背景下,高血糖可通过O-连接的β-N-乙酰葡萄糖胺化(O-GlcNAcylation)修饰等特异性分子通路,进而上调血红素氧合酶1的表达。这一特异性分子通路被证实能够显著放大氨所诱导的氧化应激与细胞衰老反应,为针对此类患者的精准干预提供了潜在的分子靶点。该通路揭示了糖尿病患者肝性脑病风险增高的新机制,也提示O-GlcNAcylation可能成为未来针对糖尿病合并肝性脑病患者的潜在治疗靶点。
氧化/硝化应激作为各种损伤因素的共同下游通路,具有重要的地位。然而,现有研究多集中于现象描述,缺乏对组织特异性和细胞特异性的氧化还原事件的深入分析。例如,星形胶质细胞与神经元内的氧化应激的差异;靶向特定细胞类型的抗氧化治疗的应用前景等。未来需进一步阐明糖尿病通过O-GlcNAcylation等特异性分子通路加剧脑内氧化应激的机制,从而为精准干预提供新的靶点。
5结语与展望
氨中毒、炎症、神经递质失衡及氧化/硝化应激4种发病机制并非彼此孤立,而是构成了一个相互交织、协同放大的复杂网络。各机制均强调肠道作为毒素与炎症主要来源的关键器官,其中氨中毒处于核心枢纽地位,其不仅直接引发星形胶质细胞水肿和能量代谢障碍,还可触发氧化/硝化应激,形成级联放大效应。各机制间协同效应显著,例如炎症与高氨血症可协同破坏血脑屏障并降低大脑对氨的耐受阈值,而氧化应激则是氨毒性和炎症损伤的共同下游机制。
从多器官交互角度总结:肝性脑病的发病过程始于肝脏与肠道,肝衰竭或门体分流导致血氨清除障碍;肠道菌群失调与屏障受损不仅导致产氨增多,还促使LPS等PAMP易位入血,继而引起多器官交互作用加剧全身紊乱,即升高的血氨和LPS激活全身免疫系统,引发系统性炎症,释放TNF-α、IL-1β及IL-6等促炎因子。上述过程受肌肉影响,肌肉作为重要的外周氨解毒器官,肌肉减少将削弱氨清除能力,从而加剧高氨血症;而肾脏在碱中毒时,铵离子排泄能力下降,进而净增加血氨负荷。最终,多器官交互汇成共同通路,导致血脑屏障破坏与脑内级联反应,使更多氨、LPS和炎症因子入脑。氨在星形胶质细胞内诱发氧化/硝化应激,导致细胞水肿和能量衰竭;炎症因子则激活小胶质细胞,放大神经炎症,破坏神经网络平衡。氧化应激作为下游通路,介导了氨和炎症因子所致的大部分细胞损伤。同时,各机制间存在冲突,临床表现无法被单一机制完全解释。例如,对于炎症与氨中毒存在“炎症为先”还是“氨毒为本”的争议:前者观点认为系统性炎症是降低大脑氨耐受阈值、破坏血脑屏障并直接损伤神经元的首要因素;后者观点认为高氨血症是启动星形胶质细胞病变和能量代谢障碍的核心始动因子。这种争议直接挑战单一靶向治疗的思路,深刻揭示针对某一发病机制的疗法在不同患者中效果有限。因此,未来的治疗策略必须转向精准分型,依据患者的主导病理机制制订联合干预方案,以期实现协同增效。
肝性脑病流行病学正经历动态转变,糖尿病和肌少症已成为重要的风险因素,通过加剧高氨血症、系统性炎症和氧化应激等多重机制参与发病,且血糖控制水平与肝性脑病风险显著相关,这为临床管理提供了新的干预靶点。
综上所述,未来研究需在多层次上整合4种发病机制所揭示的多器官交互网络,并探索糖尿病管理、肠道菌群特异性代谢产物、神经炎症的细胞特异性机制及靶向抗炎治疗等新兴领域,从而为肝性脑病的精准分型和多靶点治疗策略提供基础。肝性脑病发病机制是一个源于肝脏、累及全身、最终病于大脑的复杂网络紊乱,“多器官交互”是贯穿其中的核心主线。未来的研究与临床应用,应着眼于从细胞器、器官到整体全面探讨肝性脑病机制,推动其诊疗从对症支持迈向以重建多器官稳态为目标的治疗,最终改善患者的预后与生活质量。
https://www.lcgdbzz.org/cn/article/doi/10.12449/JCH260533
赵子昕, 祝叶青, 叶存心, 等. 肝性脑病发病机制中的多器官交互作用[J]. 临床肝胆病杂志, 2026, 42(5): 1223-1228