 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库Hepatology:安徽医科大学高潮兵等团队揭示睡眠呼吸暂停如何“伤肝”的新机理
阻塞性睡眠呼吸暂停(OSA)以反复发作的间歇性缺氧(IH)为特征,近年来被认为是代谢性疾病的重要诱因之一,包括代谢功能障碍相关脂肪性肝病(MASLD)。然而,IH 与 MASLD 之间的因果关系及其分子机制仍未完全阐明,这限制了有效治疗策略的开发。
2026年6月,安徽医科大学高潮兵,Huabing Zhang和复旦大学卫功宏共同通讯在
Hepatology
在线发表题为
KLF9 drives intermittent hypoxia-induced MASLD by suppressing the NR4A1–p38 MAPK hepatic metabolic axis
的研究论文。
该研究建立了间歇性缺氧的小鼠及细胞模型,并进行了全面的代谢与分子水平分析,包括葡萄糖耐量与胰岛素耐量实验、生化检测、组织学分析、脂质组学、实时定量 PCR、Western blot、RNA 测序、染色质免疫沉淀测序(ChIP-seq)、ChIP 实验、共免疫沉淀以及荧光素酶报告基因实验,以研究 IH 的影响及 KLF9 在肝脏脂质代谢中的作用。
结果显示,IH 暴露在正常及肥胖小鼠中均可诱导肝脏脂质沉积与胰岛素抵抗,并伴随脂质代谢通路的转录重编程。转录组分析发现,KLF9 在 IH 处理后显著上调。
功能实验表明,肝脏中过表达 KLF9 会加重 IH 诱导的脂肪变性、脂质生成及炎症反应,而敲低 KLF9 则可缓解上述变化。机制上,KLF9 可直接结合 NR4A1 启动子中的保守 GC 富集序列基序,从而抑制 NR4A1 的转录,并下调其下游 p38 MAPK 信号通路的激活。对 NR4A1 的药理学调控进一步证实,其在 KLF9 介导的 IH 相关脂质生成中发挥关键调控作用。
综上,本研究确定 KLF9 是驱动 IH 相关 MASLD 的关键转录调控因子,其通过抑制 NR4A1 并阻断 p38 MAPK 信号通路促进肝脏脂质生成。靶向 KLF9–NR4A1 轴可能为 OSA 及其他缺氧相关疾病中的 MASLD 提供新的治疗策略。

阻塞性睡眠呼吸暂停(OSA)是一种高发的慢性疾病,全球约影响近10亿人群,构成严重的公共卫生负担。其特征为睡眠过程中反复发生的上呼吸道塌陷,导致间歇性缺氧(IH)、睡眠片段化、白天过度嗜睡以及认知功能障碍。
肥胖是OSA最重要的危险因素,而OSA本身又会进一步促进多种心代谢并发症的发生,包括高血压、心血管疾病、2型糖尿病以及神经认知功能障碍。除这些全身效应外,越来越多的证据表明OSA与代谢功能障碍相关脂肪性肝病(MASLD,原非酒精性脂肪性肝病 NAFLD)密切相关,提示慢性间歇性缺氧可能加重肝脏代谢及炎症紊乱。
MASLD影响全球约四分之一的成年人。其临床谱系从单纯性肝脂肪变性到代谢功能障碍相关脂肪性肝炎、肝纤维化、肝硬化乃至肝细胞癌,是全球慢性肝病相关发病率和死亡率的重要原因之一。尽管代谢失调、胰岛素抵抗、氧化应激及炎症已被认为参与MASLD的发生发展,但其启动与进展的分子机制仍未完全阐明。
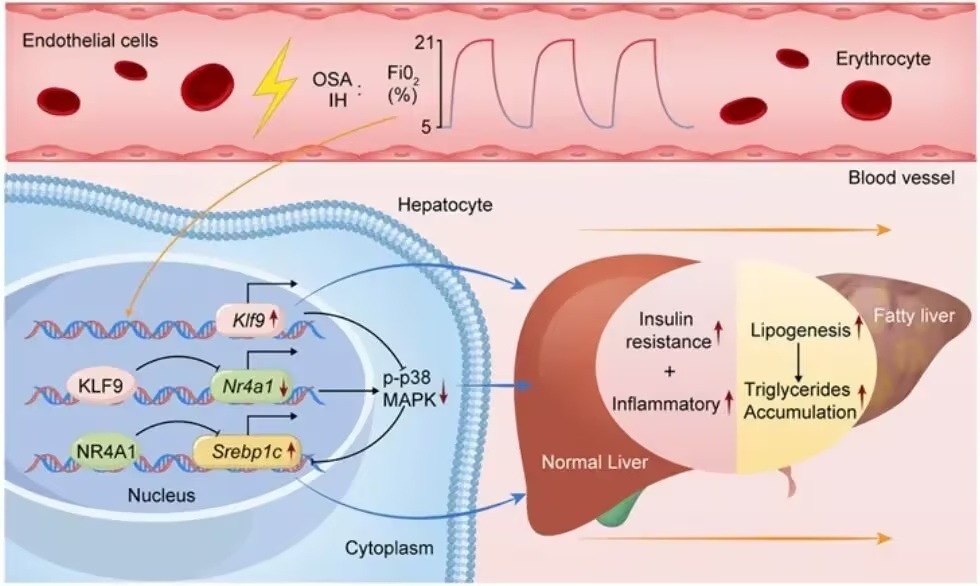
图1.全文总结图(摘自
Hepatology
)
临床及实验研究支持OSA与MASLD之间的病理生理关联。OSA严重程度与肝脂肪变性呈正相关,动物模型中间歇性缺氧可加重肝脂质沉积、炎症及胰岛素抵抗。然而,也有研究报道在某些条件下间歇性缺氧可能产生相反的保护效应,提示缺氧介导的代谢调控具有复杂性,因此亟需明确连接OSA与肝功能障碍的关键分子介质。
Krüppel样因子9(KLF9)是一种锌指转录因子,可识别GC富集序列及CACCC元件,在脂肪生成、产热、线粒体功能及氧化应激反应中发挥调控作用。既往研究提示KLF9参与葡萄糖代谢、糖异生及糖皮质激素诱导的肝胰岛素抵抗,但其在OSA相关间歇性缺氧条件下肝脂质代谢中的作用尚未明确。
本研究旨在探讨间歇性缺氧在MASLD进展中的机制作用,并鉴定KLF9作为连接OSA诱导缺氧与肝代谢紊乱的关键介质。作者发现IH诱导的KLF9通过抑制NR4A1–p38 MAPK通路,促进肝脂肪变性、炎症及胰岛素抵抗(体外及体内均证实)。药理学激活NR4A1可逆转上述效应,提示潜在治疗方向。总体而言,本研究明确了IH–KLF9–NR4A1信号轴在OSA相关MASLD中的关键作用,为疾病机制与治疗策略提供了新的理论依据。
参考消息:https://journals.lww.com/hep/10.1097/HEP.0000000000001606





