 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库Sci Adv:上海交通大学吴学锋等团队开发实体瘤CAR-T治疗新策略,靶向TMED4通过IRE1α-自噬轴增强T细胞功能
内质网应激(ERS)与自噬调控肿瘤浸润性T细胞的功能及耗竭,但其潜在机制尚不明确。
2026年6月12日,上海交通大学吴学锋,叶幼琼,北京大学曹宇和坎迪奥洛癌症研究所Christopher Heeschen共同通讯在
Science Advances
在线发表题为
Targeting TMED4 enhances CD8+ T cell function and CAR T cell efficacy in solid tumors through the IRE1α-autophagy axis
的研究论文。
该研究鉴定出ERS相关跨膜蛋白TMED4(含跨膜emp24结构域蛋白4)是CD8+ T细胞抗肿瘤免疫的关键调节因子。T细胞中Tmed4的缺失通过促进CD8+ T细胞增殖、浸润及杀伤能力增强抗肿瘤反应,同时减少终末耗竭。
机制上,Tmed4缺失过度激活肌醇需求酶1α(IRE1α)-X盒结合蛋白1(XBP1)轴,并以IRE1α依赖性方式诱导自噬流。Ern1(IRE1α)或Becn1(Beclin1)的基因缺失会削弱Tmed4缺失的抗肿瘤效应,凸显了ERS与自噬在CD8+ T细胞功能中的作用。
此外,Tmed4缺陷型嵌合抗原受体T细胞(CAR-T细胞)表现出更强的抗肿瘤免疫能力。使用反义寡核苷酸药物抑制Tmed4同样增强了CD8+ T细胞介导的肿瘤控制效果。
综上所述,本研究表明TMED4通过IRE1α驱动的自噬调控CD8+ T细胞效应功能并限制其终末耗竭,确立TMED4为改善CAR-T细胞疗效的潜在免疫治疗靶点。

CD8+ T细胞在肿瘤清除中发挥核心作用,它们通过T细胞受体(TCR)识别并杀伤癌细胞。TCR可检测主要组织相容性复合体I类(MHC-I)分子呈递的肿瘤抗原。然而,许多肿瘤通过下调MHC-I表达来逃避免疫监视,从而阻止CD8+ T细胞介导的识别和细胞毒性。
为克服这一局限性,嵌合抗原受体T细胞(CAR T细胞)疗法应运而生。CAR T细胞经工程化改造后,能以非MHC依赖的方式识别肿瘤相关抗原,使其能够在MHC-I缺失的情况下杀伤癌细胞。该疗法在治疗B细胞恶性肿瘤方面取得了显著成功,但由于免疫抑制和T细胞耗竭,其在固体肿瘤中基本无效。
肿瘤浸润性CD8+ T细胞和固体肿瘤中的CAR T细胞均存在耗竭现象,这损害了它们产生有效抗肿瘤反应的能力。T细胞耗竭的特征包括:抑制性受体的持续表达[例如,程序性死亡受体1(PD1)、T细胞免疫球蛋白和黏蛋白结构域蛋白3(TIM3)、细胞毒性T淋巴细胞相关蛋白4(CTLA4)和淋巴细胞活化基因3(LAG3)];细胞因子[干扰素-γ(IFN-γ)、肿瘤坏死因子-α(TNF-α)和白介素-2(IL-2)]分泌减少;颗粒酶B分泌降低以及增殖能力受损。
耗竭的CD8+ T细胞(Tex)存在两种不同的亚群:祖细胞样(Texprog)和终末耗竭(Texterm)T细胞。Texprog细胞(PD1int、SLAMF6+和CXCR5+)保持增殖能力、细胞因子分泌以及分化为Texterm细胞的能力。
Texterm细胞则呈现高PD1和TIM3表达,细胞溶解能力增强,但细胞因子多功能性、增殖潜力和持久性均降低。尽管转录因子,如胸腺细胞选择相关高迁移率族盒蛋白(TOX)、碱性亮氨酸拉链ATF样转录因子(BATF)、活化T细胞核因子以及信号转导及转录激活因子,调控着Tex细胞的分化,但维持T细胞耗竭并限制其抗肿瘤活性的机制仍不清楚。
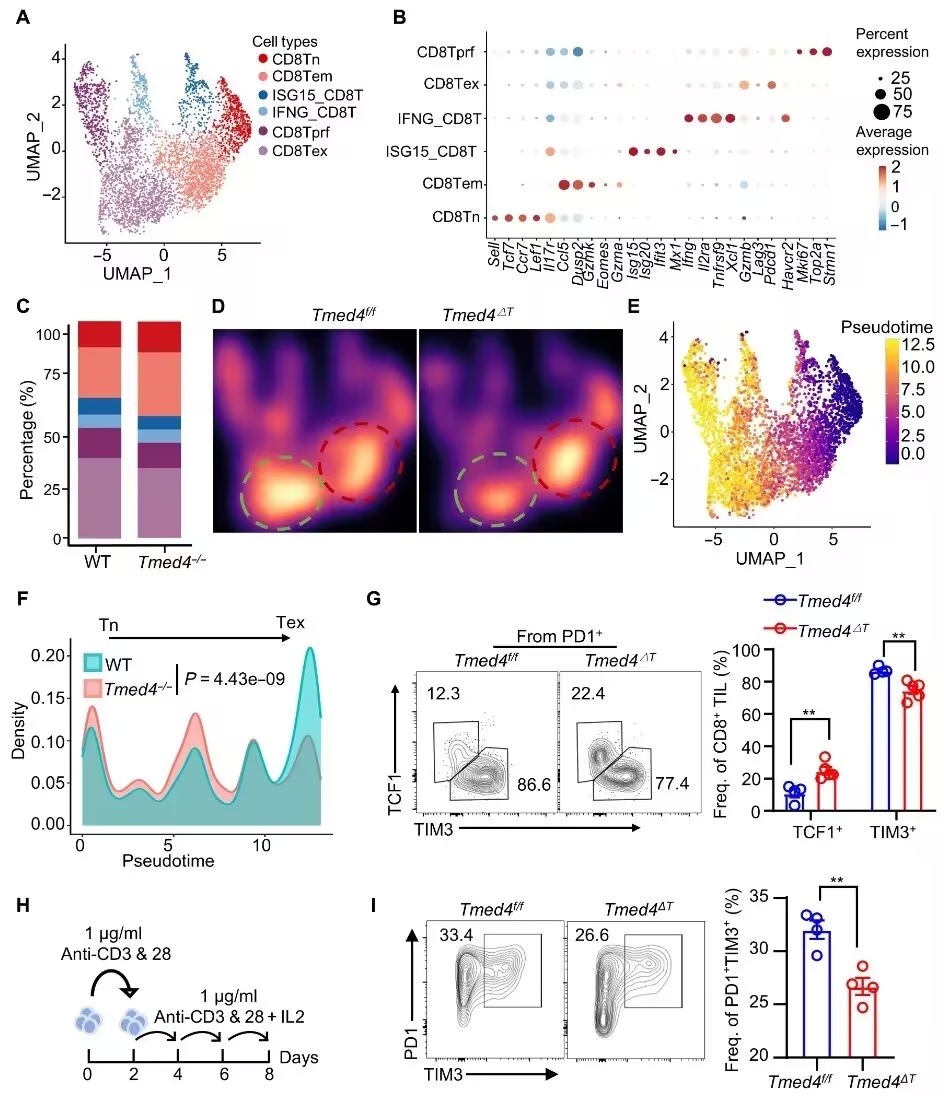
图1.Tmed4缺陷改变了CD8+ T细胞的转录谱,减少了末端衰竭(摘自
Science Advance
s)
近期研究表明,内质网应激是肿瘤微环境中T细胞抗肿瘤功能的关键调控因子。内质网应激源于未折叠和错误折叠蛋白的积累,这是高分泌细胞以及包括肿瘤、神经退行性疾病和糖尿病在内的多种疾病的标志性特征。
为恢复细胞稳态,细胞通过三种主要的内质网感应器启动未折叠蛋白反应(UPR):肌醇需求酶1α(IRE1α/Ern1)、蛋白激酶R样内质网激酶(PERK/Eif2ak3)和激活转录因子6(ATF6/Atf6)。
在正常情况下,这些感应器因与免疫球蛋白结合蛋白(BIP)结合而处于非活性状态;但在应激激活后,IRE1α将X盒结合蛋白1(Xbp1)mRNA剪接为XBP1s,PERK磷酸化eIF2α,ATF6经蛋白水解裂解为ATF6N,从而启动应激适应反应。尽管内质网应激在调节T细胞功能中的作用日益受到重视,但其在调控CD8+ T细胞耗竭中的作用仍不完全清楚。
T细胞效应功能的另一个关键内在调控因子是自噬,这是一个进化上保守的过程,细胞质组分通过溶酶体被降解。在T细胞中,自噬在TCR和细胞因子刺激后被激活,支持细胞存活、代谢适应和记忆形成。近期研究已证明,自噬能够抵抗T细胞耗竭。
例如,高细胞外K+通过限制营养摄取触发自噬以缓解耗竭,而受损的线粒体自噬则通过功能障碍线粒体的积累驱动耗竭。已知内质网应激诱导的自噬可驱动肿瘤进展和治疗耐药,但其在调控T细胞耗竭和抗肿瘤功能中的作用仍有待探索。
含有跨膜emp24结构域(TMED)的蛋白质,也称为p24蛋白,调控内质网-高尔基体运输,并与免疫调控和肿瘤进展相关。TMED4是该家族的成员之一,已被证明通过调节内质网伴侣蛋白活性和热休克因子1依赖的热休克反应来调控应激反应。
数据库分析已将TMED4表达与胶质瘤患者的免疫细胞浸润、生存和预后联系起来。作者最近证明,TMED4通过IRE1α依赖性活性氧信号和核因子E2相关因子2(NRF2)介导的抗氧化反应维持调节性T细胞(Treg细胞)功能。然而,其在CD8+ T细胞耗竭和抗肿瘤免疫中的作用仍未知。
在本研究中,作者确定TMED4是CD8+ T细胞耗竭和抗肿瘤免疫的关键调控因子。作者发现,TMED4缺失可增强CD8+ T细胞和CAR T细胞的功能,提高其抵抗耗竭和清除肿瘤的能力。机制上,Tmed4缺失激活IRE1α,导致内质网应激诱导的自噬,从而支持抗肿瘤T细胞反应。
敲除IRE1α(Ern1)或自噬调控因子Beclin1(Becn1)会消除Tmed4缺失带来的抗肿瘤益处,证实了内质网应激和自噬是调控T细胞功能的关键通路。此外,在体外和体内实验中,Tmed4缺失的CAR T细胞均表现出增强的肿瘤杀伤能力和减少的终末耗竭。
最后,作者证明,通过反义寡核苷酸(ASO)沉默TMED4可增强CD8+ T细胞的抗肿瘤能力,支持其作为改善固体肿瘤CAR T细胞疗法潜在治疗靶点的可能性。
参考消息:https://www.science.org/doi/10.1126/sciadv.aee0517





