 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库PNAS:南方医科大学顾兵等团队开发胃癌预防新工具
只有少数幽门螺杆菌(H. pylori)感染个体会沿着Correa级联进展,而经典的毒力标志物并不能完全解释这种异质性,这促使人们采用全基因组方法来量化与晚期病变相关的菌株水平基因组风险。
2026年6月9日,南方医科大学顾兵,Liang Wang和四川大学Barry J. Marshall共同通讯在
PNAS
在线发表题为
Risk prediction of Helicobacter pylori strains across Correa’s cascade via intelligent analysis of genome-wide SNPs
的研究论文。
该研究收集了528个高质量幽门螺杆菌全基因组序列,涵盖非萎缩性胃炎(NAG)、萎缩性胃炎(AG)、肠上皮化生(IM)和胃癌(GC),并在明确校正群体结构后进行了细菌全基因组关联分析。
然后,作者将与晚期病变相关的变异整合到一个随机森林模型中,以推导出幽门螺杆菌基因组风险评分(HpRS),该评分定义为菌株与晚期病变(IM/GC)而非非晚期病变(NAG/AG)相关联的预测概率。
尽管系统发育分析显示全基因组聚类主要受地理谱系而非疾病分期驱动,但HpRS在重复交叉验证中表现出强大的区分能力(平均AUC = 0.902,95% CI: 0.892–0.912),在内部保留集中仍具有区分能力(AUC = 0.780),并推广到两个独立队列(细菌与病毒生物信息学资源中心(BV-BRC):AUC = 0.871;医院队列区分IM与NAG/AG:AUC = 0.843)。
具有预测作用的单核苷酸多态性主要映射到核心功能(DNA修复、翻译、中枢代谢和脂质代谢),并且通常影响保守结构域,这表明存在多基因结构并产生了可测试的功能假设。HpRS为考虑菌株的风险分层提供了一个概念验证框架,并可能补充未来的胃癌预防策略。
胃癌仍然是全球癌症相关死亡的主要原因之一,并且幽门螺杆菌(H. pylori)感染已被确定为其主要病因因素。幽门螺杆菌相关肠型胃癌的发生通常由称为Correa级联的经典多步序贯模型来描述。
在该模型中,慢性幽门螺杆菌感染驱动正常胃黏膜通过一系列癌前病变(包括非萎缩性胃炎(NAG)、萎缩性胃炎(AG)、肠上皮化生(IM)和异型增生)进行转变,最终发展为胃癌(GC)。
在这些阶段中,进展到IM被广泛认为是一个重要的临床和生物学转变,反映了胃黏膜的显著重塑和恶性转化风险的增加。基于这些明确界定的组织学阶段,Correa级联提供了一个生物学上连贯的框架,将慢性感染与逐步的胃癌发生联系起来。
尽管幽门螺杆菌在全球范围内流行率很高,但只有一小部分感染者最终发展为胃癌。越来越多的证据表明,临床异质性不能仅用宿主或环境因素来解释,这凸显了幽门螺杆菌自身毒力因子和遗传多样性对疾病进展的关键贡献。
这些观察结果强调了需要系统地研究细菌基因组多样性如何影响Correa级联中的不同转变,旨在定义与晚期病变相关的菌株水平基因组信号,并为未来的风险分层框架提供信息。
幽门螺杆菌菌株表现出显著的遗传异质性,其致癌潜力也不相同。经典地,幽门螺杆菌毒力是通过一组典型因子进行研究的,包括cag编码的IV型分泌系统和CagA癌蛋白、VacA细胞毒素、BabA黏附素以及多种外膜蛋白。
这些标志物在人群水平上与炎症增强、黏膜萎缩和胃癌风险增加相关,并为作者对发病机制的理解提供了信息。然而,这些毒力因子的存在或缺失本身并不能完全解释在感染者中观察到的广泛疾病结局谱。最近的研究表明,特定的细菌单核苷酸多态性(SNP)可以发挥深远的功能效应,并在胃癌发生中起关键作用。
里程碑式的研究已经在关键细菌基因中鉴定出与癌症相关的SNP,包括CagA磷酸化基序内的多态性以及丝氨酸蛋白酶HtrA中的多态性,这些多态性直接调节宿主炎症信号、破坏上皮连接完整性并诱导DNA双链断裂。总的来说,这些发现表明核苷酸水平的遗传变异是幽门螺杆菌相关癌症风险的关键决定因素。
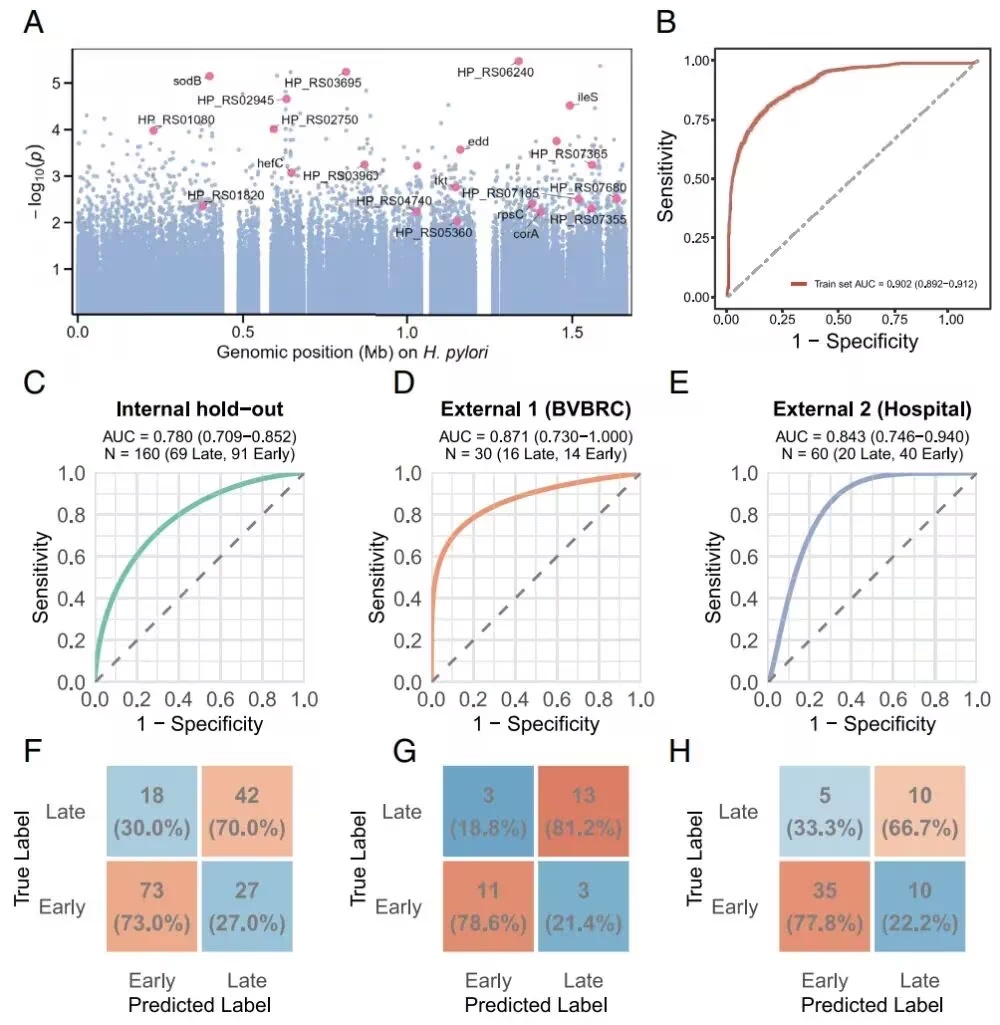
图1.疾病分期相关SNP的全基因组关联分析(摘自
PNAS
)
与此同时,全基因组测序(WGS)研究已经记录了全球不同分离株中幽门螺杆菌群体结构和泛基因组内容的丰富多样性。然而,迄今为止,大多数大规模基因组分析都集中在群体遗传学、系统地理学或抗生素耐药性上,而不是疾病分期特异性的致病性适应。
因此,使细菌能够长期持续存在并主动驱动从良性胃炎向晚期癌前病变和恶性肿瘤进展的细菌遗传变化仍然未被完全阐明。因此,解读菌株特异性基因组变异如何影响Correa级联中不连续的组织学转变,对于将描述性细菌基因组学转化为可操作的精准风险分层至关重要。
在此,作者假设幽门螺杆菌的致癌性由菌株特异性基因组变异所支配,这些变异共同促进其超越可逆性胃炎进展为不可逆的癌前和恶性状态。为了解决既往候选基因方法的局限性以及对菌株水平基因组分层的迫切需求,作者收集了涵盖Correa级联四个关键阶段的幽门螺杆菌基因组集合。
作者应用了细菌全基因组关联研究(GWAS)框架,严格控制系统发育混杂因素,以系统性地鉴定与疾病分期对比相关的SNP。作者进一步使用机器学习方法整合这些变异,最终开发了基于随机森林(RF)的幽门螺杆菌基因组风险评分(HpRS)。
该评分定量估计给定菌株与晚期(IM/GC)病变 vs 非晚期(NAG/AG)病变相关联的概率,从而将复杂的细菌基因组变异转化为一个单一的、可解释的菌株水平基因组评分。
作者进一步评估了HpRS在不同系统发育谱系中的可移植性,并检查了模型选择的特征是否显示出超出谱系背景之外的共享方向性模式。通过重复交叉验证(CV)严格评估模型性能,并在独立队列中进行验证,同时对关键贡献的SNP进行功能注释以生成可测试的生物学假设。
总之,作者的工作将癌症相关细菌变异的研究从孤立的候选基因座扩展到了一个全基因组的、系统水平的框架。通过在Correa级联的背景下描绘与晚期病变相关的集体基因组特征,作者为菌株水平的基因组分层提供了概念验证方法,对未来的胃癌预防策略具有潜在的相关性。
参考消息:https://doi.org/10.1073/pnas.2603926123





