 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库Autophagy:山东大学侯宇团队发现免疫性血小板减少症治疗新策略
干扰素γ(IFNG)激活的巨噬细胞在免疫性血小板减少症(ITP)中加速血小板的清除并促进炎性细胞因子的过度分泌。
2026年6月26日,山东大学侯宇独立通讯在
Autophagy
在线发表题为
Chaperone-mediated autophagy reprograms heterogeneous subsets of activated macrophages in immune thrombocytopenia
的研究论文。
该研究识别出两种不同的活化巨噬细胞亚群:以吞噬活性为主的吞噬性巨噬细胞(M[IFNG]p)和以促炎能力为特征的炎性巨噬细胞(M[IFNG]i)。在ITP患者中,M(IFNG)p和M(IFNG)i亚群的功能发生紊乱。
研究发现ITP中巨噬细胞的分子伴侣介导的自噬(CMA)存在缺陷,并已有报道指出该缺陷可诱导持续性炎症。此外,体外实验表明,CMA干扰增强了巨噬细胞的促炎功能,而非其吞噬活性。
与此一致,巨噬细胞特异性条件性敲除溶酶体相关膜蛋白2A(lamp2a)的小鼠显示出M(IFNG)i亚群的扩增及细胞因子的过量释放。在ITP中,恢复CMA可通过醛脱氢酶2家族成员(ALDH2)抑制M(IFNG)i亚群并重编程M(IFNG)p亚群。
本研究将抗整合素β3(ITGB3)/CD61免疫致敏的脾细胞转移至重症联合免疫缺陷小鼠体内,建立ITP活性小鼠模型。CMA激活降低了活化巨噬细胞的促炎和吞噬活性,并改善了ITP小鼠的血小板减少症。通过MeRIP测序,鉴定出磷酸酶和张力蛋白同源物(PTEN)是LAMP2A的关键激活因子,其m6A甲基化水平降低并继发下调,这提示了活化巨噬细胞中CMA缺陷的潜在机制。
总之,在ITP中,恢复CMA通过ALDH2抑制M(IFNG)i亚群并重编程M(IFNG)p亚群,从而提高血小板计数。针对原发性活化人巨噬细胞中的CMA进行治疗,为快速且持续地提高ITP血小板计数并恢复免疫平衡提供了一种有前景的治疗策略。

巨噬细胞在自身免疫性疾病的发病机制中发挥双重作用:既作为效应细胞,也作为抗原呈递细胞。巨噬细胞的激活需要干扰素-γ(IFNG)作为启动信号,这有助于促进病原体识别、抗原处理与呈递、抗病毒状态以及免疫调控的上调。
IFNG刺激模拟了辅助性T(Th)1细胞或T-bet细胞对单核细胞和巨噬细胞的激活作用。Decano JL等人通过单细胞RNA测序,鉴定了IFNG诱导的巨噬细胞的两个不同亚群:分别被命名为炎性M(IFNG)i亚群和吞噬性M(IFNG)p亚群。M(IFNG)i亚群主要表现为促炎细胞因子的产生增加,而M(IFNG)p亚群则表现出增强的吞噬作用、胞葬作用和趋化能力。
激活的T细胞产生的IFNG可诱导单核细胞/巨噬细胞中信号转导及转录激活因子(STAT)的激活,从而产生更高水平的趋化因子和细胞因子,这加剧了系统性红斑狼疮的病情发展。在原发性免疫性血小板减少症(ITP)中,巨噬细胞导致抗体介导的血小板过度吞噬。此外,在ITP中,IFNG诱导的STAT1核转位促进了巨噬细胞的吞噬活性。
ITP是一种自身免疫性出血性疾病,其特征为网状内皮系统对血小板的清除加速。已有研究表明,单核细胞和巨噬细胞(其特征为内质网应激通路富集,以及CD44和肿瘤坏死因子-α(TNF-α)水平升高)在ITP的发病机制中起关键作用。
作者先前发现,血小板生成素(THPO)受体激动剂可通过下调单核细胞上激活型Fcγ受体(FCGR/FcγR)的表达并上调抑制型FCGR的表达,来减少ITP患者及小鼠模型中的血小板破坏。此外,据报道,乙醛脱氢酶2家族成员(ALDH2)是巨噬细胞胞葬作用内化过程所必需的,这可能与M(IFNG)p的功能相关。
然而,ITP患者中强烈的吞噬活性是否归因于激活的巨噬细胞的特定亚群,以及ITP患者中偏向Th1谱的细胞因子模式和更高水平的IFNG是否有利于M(IFNG)p和M(IFNG)i的分化及生物学功能,这些问题仍有待阐明。
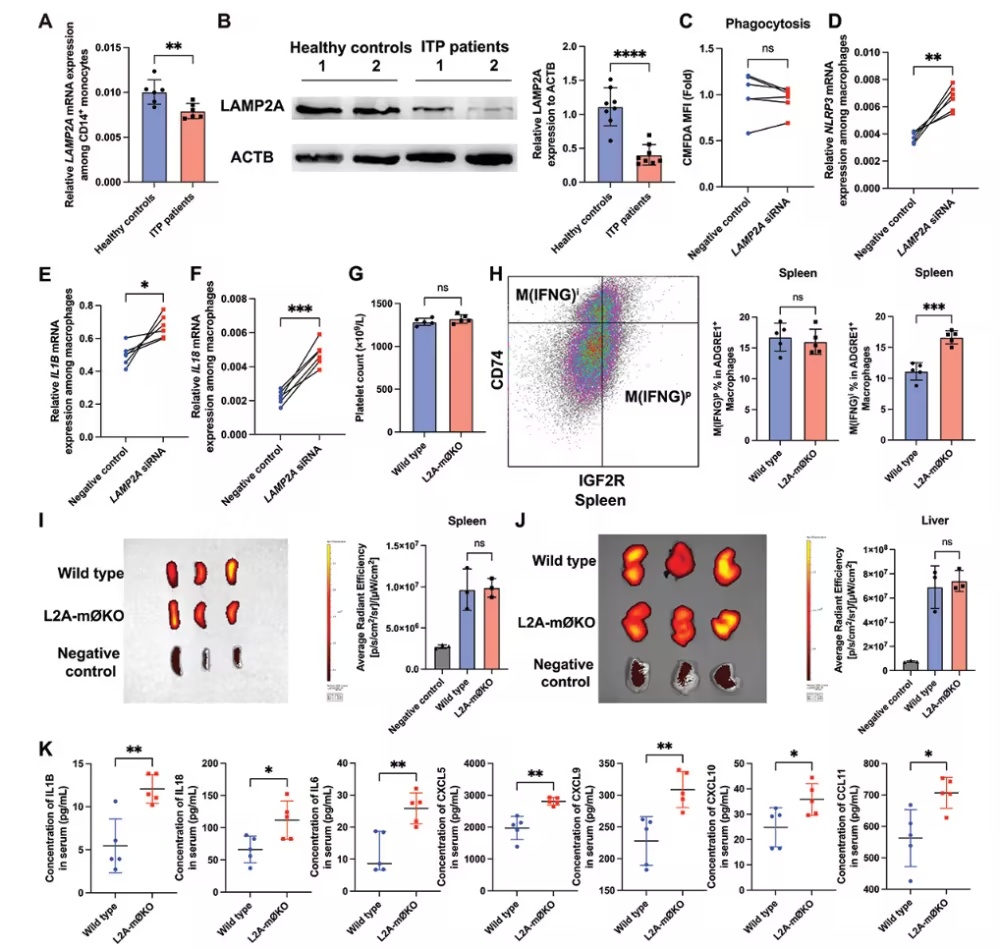
图1.CMA的下调增强了巨噬细胞的促炎功能,而不是吞噬活性(摘自
Autophagy
)
巨噬细胞表型极化是一个受多种信号通路调控的复杂动态过程。自噬是将靶蛋白运输至溶酶体进行降解的过程。分子伴侣介导的自噬(CMA)是自噬的一种特殊形式,它选择性地降解含有KFERQ基序的底物。
CMA底物在溶酶体中被分子伴侣标记,以调节细胞的关键功能,如代谢通路的调控、细胞周期的控制以及免疫应答。溶酶体相关膜蛋白2A(LAMP2A)在CMA中起着关键作用。LAMP2A的水平直接决定CMA的活性。在间充质基质细胞中,反映IFNG和TNF-α的作用,CMA受到抑制,表现为LAMP2A表达下降。
从机制上讲,CMA的抑制显著促进了IFNG联合TNF-α诱导的NFKB和STAT1的激活,从而导致间充质基质细胞中C-X-C基序趋化因子配体10(CXCL10)和诱导型一氧化氮合酶2(NOS2/iNOS)的表达增强。在非酒精性脂肪性肝炎中,单核细胞来源的巨噬细胞中的CMA受损。
CMA缺陷促进了非酒精性脂肪性肝炎小鼠的巨噬细胞浸润、脂肪性肝炎和纤维化。在动脉粥样硬化小鼠模型中,巨噬细胞中CMA的缺陷导致NLR家族 pyrin 结构域包含蛋白3(NLRP3)炎症小体的持续激活,从而促进血管炎症和动脉粥样硬化进展。然而,在ITP中,CMA与激活的巨噬细胞之间的关联尚未得到明确阐释。
先前的研究报道,热休克同源蛋白70(CMA中的一种必需分子伴侣)的表达与甲基转移酶3,N6-腺苷酸甲基转移酶复合物催化亚基(METTL3)-METTL14依赖的N6-甲基腺苷(m6A)甲基化显著相关。
在本研究中,甲基化RNA免疫共沉淀测序(MeRIP-seq)揭示,磷酸酶和张力蛋白同源物(PTEN,CMA的一个关键激活因子)表现出m6A甲基化水平降低及其继发的表达下调。然而,在ITP中,METTL3是否通过PTEN的m6A修饰来调控CMA尚不清楚。
此外,尽管已知ALDH2协调自噬的多个阶段,包括通过磷酸化AMP活化蛋白激酶(AMPK)和抑制雷帕霉素靶蛋白激酶(MTOR)激活来启动自噬、通过上调并释放BECN1(beclin 1)来促进自噬泡成核、以及通过损害溶酶体功能来影响自噬体与溶酶体融合,但ALDH2是否在CMA中也发挥作用仍然未知。
本研究的目的是阐明ITP患者激活的巨噬细胞中CMA受损的潜在分子机制。在此,作者研究了CMA水平,并检测了LAMP2A对促炎性巨噬细胞和吞噬性巨噬细胞功能的影响。利用转录组和甲基化测序技术,作者探究了其潜在的分子机制。此外,作者使用了活动性ITP小鼠模型来进一步验证LAMP2A是否可作为潜在治疗靶点。
参考消息:https://doi.org/10.1080/15548627.2026.2693777





