改良药“伪创新”退潮:挤干剂型套利泡沫,回归真实技术硬核
发布时间:2026-07-16来源:药事纵横
在国内医药创新转型的这几年,改良型新药(2类新药)被市场过度神化了。业内普遍流传着一套固定认知:依托成熟上市药物、无需验证全新安全性、研发周期短、投入低、容错率高,是中小药企摆脱仿制药内卷、快速切入创新赛道的最优解。也正是基于这套认知,过去五六年,国内药企扎堆布局剂型微调、规格改动、给药形式调整的轻改良项目,一度让改良药管线成为行业扩容最快的板块。但从我近几年跟进的数十个2类新药申报项目、CDE审评反馈及大量落地失败案例来看,行业对改良药的认知存在严重偏差。很多企业误以为“改个剂型就是创新”,盲目跟风立项,最终要么卡在临床试验阶段,要么拿到BE合格结果却倒在注册核查,还有不少品种上市后因为无临床差异化优势,直接面临集采降价、滞销亏损的局面。看似门槛极低的赛道,实则暗藏大量隐性审评壁垒和产业化陷阱。一、研发分层并非理论分类,而是项目落地与盈利的真实分水岭
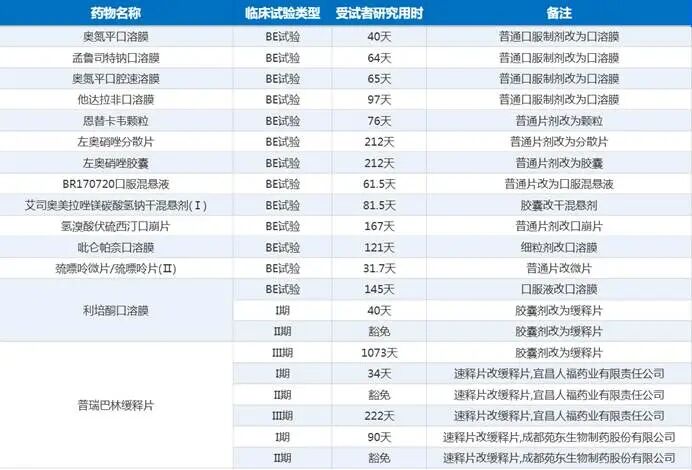
市面上多数行业稿件会机械地按照剂型类型划分改良药等级,但这种纯理论分类,完全脱离企业立项和商业化实操。在实际研发与申报过程中,改良项目的核心差距,从来不是剂型形式的差异,而是是否改变药物PK行为、是否具备可量化的临床获益、是否构建技术壁垒。基于一线项目落地经验,改良药可以清晰分为三个层级,不同层级的研发难度、周期、审评通过率、盈利空间天差地别,这也是行业两极分化的根本原因。第一类为无PK改变的剂型适配改良,也是目前行业泡沫最严重的板块。这类项目以普通片剂、硬胶囊、颗粒剂、分散片的互相替换,肠溶制剂剂型微调为核心,全程不改变药物体内吸收、分布、代谢、排泄特征,PK曲线与原研参比制剂基本一致,仅需完成生物等效性试验即可满足申报基础要求。但实操中这类项目几乎没有长期价值,也是行业踩坑重灾区。最典型的公开案例就是某药企阿莫西林分散片改良项目(受理号:CXHL2200XXX),2022年启动BE试验,2023年顺利完成临床试验并提交注册申请,全程研发流程顺畅,无技术卡点。但该品种申报阶段,国内已有近30家企业获批同类剂型,产品无任何临床差异化优势,仅实现剂型补充。2024年获批上市后,直接纳入集采竞价,中标价格大幅低于研发成本,企业完全无法回本。这类项目的共性问题非常明确:极速落地、零技术壁垒、同质化泛滥,本质是无效管线填充,不属于真正的医药创新。第二类为PK优化型缓控释剂型改良,是目前性价比最高、审评最稳的中端创新赛道。针对普通速释制剂服药频次高、血药浓度峰谷波动大、胃肠道不良反应频发的临床痛点,通过骨架缓释、薄膜包衣、渗透泵等制剂技术,调整药物释放速率,平稳血药浓度、降低毒副作用、提升患者用药依从性。因为直接改变药物PK/PD特征,体内药代动力学行为发生实质性变化,监管层面不认可单纯BE等效,必须开展相对生物利用度研究,同时设计针对性临床试验,量化验证改良后的临床优效性。这类项目研发周期普遍在1—3年,对制剂工艺、处方筛选、体内外相关性(IVIVC)研究要求较高,中小药企技术储备不足,布局量相对偏少,但落地质量极高。国内多款慢病长效制剂,如降压、降糖类缓释剂型改良品种,均凭借可量化的依从性优势、安全性优化优势,顺利通过审评、纳入医保,避开低端内卷,实现稳定商业化放量,是目前最贴合临床需求、风险收益比最优的改良方向。第三类为复杂注射剂系统性改良,是改良药赛道的硬核高壁垒领域,研发难度无限接近1类创新药。将普通口服制剂、常规水溶注射剂改造为长效微球、纳米脂质体、靶向缓释注射剂,需要突破精细制剂工艺、无菌生产、质量稳定性控制、体内靶向递送等核心技术。根据《中国医药研发蓝皮书(2026)》临床试验样本数据,该类项目临床试验周期区间为615天—1556天,原则上需要完整完成I、II、III期递进式临床试验,豁免临床的审批门槛极高,全年获批豁免案例不足5%。这类项目不存在同质化竞争问题,头部企业布局的长效注射剂品种,上市后可长期独占细分赛道,不受集采低价冲击。但必须客观承认其短板:研发投入动辄数千万元,生产放大难度大,工艺稳定性容错率极低,不少头部企业的在研项目,均卡在生产工艺放大和III期临床安全性核查阶段,并非所有高端改良项目都能顺利落地,行业不能盲目追捧复杂制剂赛道。图1. 口服用药之间的改剂型二、优先审评利用率低迷,核心是伪创新泛滥挤占审评资源
很多研发从业者在实操中都会吐槽:改良药创新难做、政策红利难拿。从2017—2025年CDE公开审评统计数据来看,这一感受确实有数据支撑。2025年全年改良型新药申报上市品种中,仅5个品种纳入优先审评,占比仅7%;全年6个优先审评获批品种,占年度获批总量的17%,优先审评通道的利用率长期处于低位。行业普遍将其归咎于审评标准严苛,但结合大量被否、被驳回的项目审评意见来看,核心问题从来不是政策收紧,而是行业无效、低效的伪创新项目过多。CDE优先审评的核心准入标准,始终锚定“临床未满足需求、可量化临床获益”,但当前行业九成以上的改良项目,仅做剂型、规格、包装的表层调整,既不能解决特殊人群用药难题,也无法优化药效、降低不良反应,不存在任何实质性临床价值。这里也能解释行业数据看似矛盾的问题:公开数据显示改良型新药九年平均审评时长为467天,但优质合规项目的审评周期大多集中在201—400天。数据偏差的核心原因是,海量低端劣质项目存在申报资料不规范、临床论证缺失、试验设计不合理等问题,需要多次补正、现场核查、答疑修正,大幅拉高了行业整体平均审评时长。优质、具备真实临床优势的项目,审评效率远高于行业均值,不存在普遍的审批卡点。业内长期存在两极争议,也能印证当前行业乱象。中小药企普遍认为,改良药作为中小企业创新突破口,应当适度放宽审评容错空间,给予试错机会;而审评机构、头部研发企业则坚持,必须严格甄别改良创新质量,避免大量无价值项目挤占公共审评资源、扰乱创新秩序。结合近两年审评态势来看,监管端的态度已经非常明确,未来只会持续收紧低端改良项目的审批标准,伪创新的生存空间会持续压缩。三、改良药“易研发、难获批”悖论:不止是人为问题,技术短板同样突出
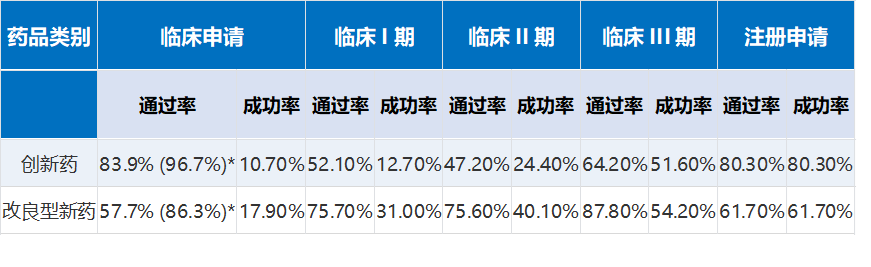
行业一直存在一个经典数据悖论,也是很多企业立项踩坑的关键:改良型新药整体研发成功率17.9%,高于小分子原创药的10.7%,看似风险更低,但细分审评通过率全面落后。其中改良药IND批准率86.3%、临床申请通过率57.7%、注册通过率61.7%,而原创创新药对应数据分别为96.7%、83.9%、80.3%。图2. 中国化学创新药和改良型新药研发的通过率与成功率(*为临床申请的CDE批准率)很多行业分析会简单二元对立,认为“原创药失败是技术问题,改良药失败是企业套利思维、论证不足的人为问题”,但结合一线实操来看,这个结论过于片面,存在明显的逻辑局限性。改良药审评失利,除了企业重工艺、轻临床、论证敷衍的主观问题,同样存在大量客观技术短板。最典型的案例为某企业盐酸氨溴索口崩片改良注册驳回案例,该项目顺利完成BE试验,工艺稳定、质量合格,但注册阶段被CDE驳回。审评意见明确指出:项目仅完成剂型形态改良,未针对吞咽障碍患者、儿童患者开展针对性临床研究,无法证明口崩剂型相较于普通片剂的临床适配优势,改良无实质性价值。这是典型的“人为论证缺失”导致的失败,也是多数低端改良项目的通病。但同时,大量中高端改良项目的失利,完全源于技术壁垒不足。不少企业布局缓控释制剂改良项目,前期处方筛选不完善,生产放大过程中出现释药曲线不稳定、批间差异大、辅料相容性不达标等技术问题,即便临床试验勉强完成,也无法通过审评核查。复杂注射剂项目更是普遍存在无菌控制难、制剂稳定性差、体内释放不可控等技术卡点,这类失败和企业套利思维无关,纯粹是国内制剂技术积累不足导致的客观短板。除此之外,行业严重低估了改良药的成本分层风险。根据公开研发投入样本数据,基础剂型改良临床前投入仅100万元起,临床试验投入数百万元;但新适应症拓展、复杂注射剂改良项目,临床前投入最高可达2000万元,临床试验投入区间高达3000万—6亿元。很多中小企业盲目跟风高端赛道,资金、技术、团队均不匹配,临床试验草草收尾,最终项目报废,造成严重的资源浪费。四、行业去泡沫不是淘汰改良赛道,而是淘汰低端套利模式
复盘国内改良型新药近十年发展,赛道已经完整经历了野蛮生长、产能过剩、泡沫初现的完整周期。行业发展初期,国内药品剂型供给不完善、特殊人群用药缺口大,89%的基础剂型改良品种,确实能够补充市场空白、完善用药体系,具备阶段性产业价值。但随着医药产业升级、集采常态化、医保支付精细化,单纯补全剂型的低端改良模式,已经完全不符合当前产业发展需求,行业去泡沫化是必然趋势。结合2026年最新修订的《药品注册管理办法》配套细则、医保双轨支付政策、集采提质稳价导向来看,未来改良药行业的升级逻辑,并非全面否定改良创新,而是彻底淘汰无价值的套利式改良,倒逼行业回归临床真实需求。低端同质化改良项目会持续出清,审评核查的重点将全面聚焦“临床优效性”,没有可量化获益的剂型微调项目,立项价值基本归零。未来企业立项,必须紧扣特殊人群适配、用药安全性优化、依从性提升、新适应症拓展等真实临床痛点,依托真实世界研究数据支撑改良价值,彻底摆脱跟风扎堆的粗放模式。制剂技术壁垒将成为核心竞争核心,行业竞争从“拼速度、拼数量”转向“拼工艺、拼技术”。传统普通固体制剂、常规注射液的简单改良逐步退场,缓控释渗透泵技术、靶向递送、纳米制剂、长效微球等高端制剂技术,成为头部企业的核心护城河。2.1类结构改良、2.3类复方改良等高壁垒品种的申报占比持续提升,推动改良药从表层剂型改动,转向实质性技术创新。商业化体系的完善,也在重塑行业竞争格局。医保NDA预申报机制落地,大幅缩短了改良药上市后的准入周期;医保与商业保险双联动,解决了高端改良药价格偏高、支付受限的难题。同时,集采摒弃单纯低价导向,侧重质量与价值筛选,让优质改良药避开恶性价格内卷,叠加院外零售、互联网医疗渠道扩容,优质品种实现全域放量,行业资源持续向技术、资金、合规能力更强的头部企业集中,中小药企的生存空间进一步收窄。需要理性看待的是,国产改良药出海目前仍处于试水阶段,不能过度夸大国际化进度。国内高端改良制剂技术虽逐步成熟,但海外注册壁垒、国际市场渠道、海外临床认可度仍存在明显短板,短期无法成为行业主流增量,国际化布局仍是长期深耕方向。五、结语
站在2026年的产业节点回看,改良型新药赛道的最大问题,从来不是创新门槛过高,而是行业长期存在的认知错位和套利思维。很多企业将改良药当成创新捷径,忽视了审评合规要求、临床价值本质和技术壁垒短板,盲目扎堆低端改良,制造了大量行业泡沫,也导致赛道口碑两极分化。必须认清的是,改良型新药是精细化、高合规要求的创新赛道,而非低成本套利工具。未来行业的良性发展,需要摒弃模板化的创新思维,不再追求形式上的剂型改动,而是立足临床真实未满足需求,依托扎实的制剂技术、严谨的临床研究、完善的合规体系,做有价值、有壁垒、有商业化潜力的真改良创新。只有淘汰伪创新、深耕真技术,国内改良型新药产业才能彻底完成去泡沫升级,实现从剂型补充到技术创新的实质性跨越。[2] 王卓,李詠恩.国内外改良型新药的注册评价政策及其实际临床价值评价[J].中国处方药,2026,24(4):1-6.[3] 朱海健.改良型新药的前沿技术与开发策略[J].药学进展,2025,49(3):161-163.立即扫码加入药事纵横交流群
转载说明:本文系转载内容,版权归原作者及原出处所有。转载目的在于传递更多行业信息,文章观点仅代表原作者本人,与本平台立场无关。若涉及作品版权问题,请原作者或相关权利人及时与本平台联系,我们将在第一时间核实后移除相关内容。
 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库