 五度妙笔
五度妙笔 API商城
API商城
 数据库
数据库CIRS发布六大药监机构新药注册效率分析年报

*题图仅作示意用
7月初,监管科学创新中心(Centre for Innovation in Regulatory Science, CIRS)发布了其年度分析报告,聚焦于2016至2025年间EMA、FDA、日本药品和医疗器械管理局(Pharmaceuticals and Medical Devices Agency, PMDA)、加拿大卫生部(Health Canada,HC)、瑞士医药管理局(Swissmedic)以及澳大利亚治疗产品管理局(Therapeutic Goods Administration, TGA)六家监管机构对新活性物质(New Active Substance, NAS)的批准情况。
以下内容编译自报告的“关键信息”部分,并从中国药监《2025年度药品审评报告》中摘出部分相关数据作为对比参考。完整报告(及往年报告)请至官网或识林查阅。此外,CIRS从2025年开始将中国药监纳入统计范围,整合报告可能稍晚推出。
数量:近5年更多新药获得全球上市
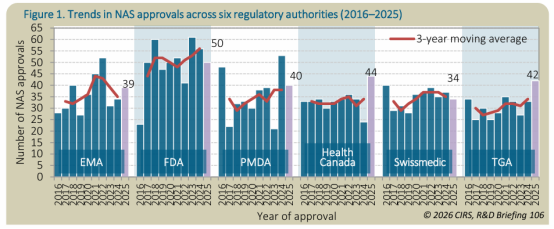
*批准数量总览
2025年,FDA 批准的新活性物质(NAS)数量最多(50个),其次是加拿大卫生部(44个)、TGA(42个)、PMDA(40个)、EMA(39个)和 Swissmedic(34个)。FDA 在过去十年中批准的 NAS 数量多于其他同类监管机构。但并非所有产品都得到了及时的全球化注册申报。
*NMPA全年批准上市 1 类创新药 76 个品种。
被所有六个监管机构共同批准的 NAS 数量从 2016-2020 年的 37个 增加到 2021-2025 年的 52个。
速度:日本PMDA在努力提速,更多利用加速机制
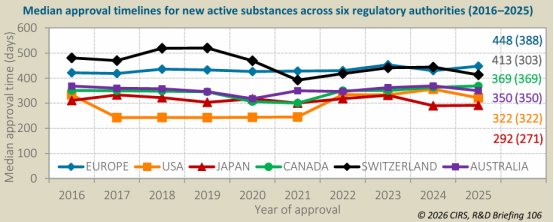
*批准时间中位数
2025年,PMDA 的审批时间中位数最短(292天),其次是 FDA(322天)、TGA(350天)、加拿大卫生部(369天)、Swissmedic(413天)和 EMA(448天)。
审评类型对审批速度的影响
2025年,所有六个监管机构的快速审评时间中位数均明显短于标准审评。EMA 和加拿大卫生部的快速审评时间中位数最短(均为 235天),而 TGA 最长(306天)。EMA 的审评类型差异最大,快速审评比标准审评快 217天。
快速审评路径使用率
各机构对快速审评路径(如加速审评、优先审评等)的使用率存在显著差异。最高的是 FDA(56%),其次是 PMDA(53%)、加拿大卫生部(25%)、Swissmedic(18%)、TGA(10%)和 EMA(5%)。PMDA 的快速审批比例在 2025 年从上年的 34% 大幅增加至 53%。在 2021-2025 年期间,除 FDA 外,各机构对快速路径的使用率基本保持稳定,而 FDA 呈现逐年下降趋势。
监管促进路径的应用
与 2016-2020 年相比,2021-2025 年间所有机构对监管促进路径(简称FRP,包括FDA快速通道和突破性疗法等,报告P21有清单介绍)的使用均有所增加。其中,FDA 使用最频繁(75% 的批准涉及至少一种 FRP),其次是 Swissmedic(68%)、TGA(62%)、加拿大卫生部(55%)、PMDA(42%)和 EMA(36%)。
附条件与临时批准路径
2025年,通过条件性/临时/暂时路径批准的 NAS 比例分别为:FDA 24%、EMA 23%、加拿大卫生部 16%、Swissmedic 6%、TGA 5% 和 PMDA 3%。
FDA 审评周期与重大增补
2025年,FDA 批准的 NAS 中有 74% 是在首个审评周期内获批的,为近五年来的最高比例。第一周期且无重大增补(major amendments)的批准时间最短(中位数 243天),而经历多周期才获批的产品,其审批时间通常会超过 500天。
不同治疗领域的审批速度
在 2021 至 2025 年期间,抗感染药物在六个监管机构中的整体审批时间中位数最短(273天),其次是消化及代谢药物(328天)、抗肿瘤及免疫调节剂(353天)、血液系统药物(365天)以及神经系统药物(414天)。
*NMPA的76个1类创新药中,26 个品种(34.2%)通过优先审评审批程序批准上市,15 个品种(19.7%)附条件批准上市,15 个品种(19.7%)在临床试验期间纳入了突破性治疗药物程序。
孤儿药认定和审批效率
2025年,获得孤儿药认定的 NAS 批准比例在各机构均处于高位,其中 Swissmedic 最高(62%),其后为 FDA(54%)、PMDA(53%)、EMA(38%)和 TGA(36%)(图12)。2025年,PMDA 的孤儿药审批中位数最短,为 269天,其次是 FDA(322天)、TGA(373天)、EMA(452天)和 Swissmedic(452天)。
*NMPA批准的1类创新药中,罕见病药物6个品种,占比约8%。
申报间隔:美欧同步是主流,大药企进入日本相对迟缓
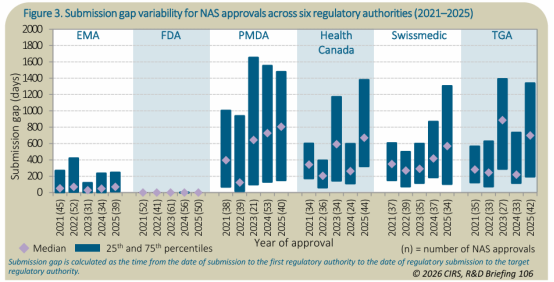
*申报间隔总览
随着药企更多寻求全球多个市场批准,申报序列发生了转变,PMDA 的递交时间有所提前,这也反衬加拿大卫生部、Swissmedic 和 TGA 的申报间隔中位数变大。
申报间隔与变异性
2025年,以FDA 的申报间隔中位数作为基准(0天),其次是 EMA(69天)、Swissmedic(569天)、加拿大卫生部(670天)、TGA(700天)和 PMDA(807天)。各机构间申报间隔的变异性也存在显著差异,四分位距(IQR)从 0天(FDA)到 1326天(PMDA)不等。新药在日本申报的滞后时间差异极大,部分药物在海外获批多年后才在日本递交上市申请。EMA 的 IQR 在六家机构中第二窄,为 229天,反映出新药引入欧洲市场的时间预期高度一致且可预测,美欧同步仍是主流。
企业规模与申报策略
2025年,对于大多数监管机构,非百强制药企业的申报间隔中位数普遍长于百强制药企业。其中差异最大的是加拿大卫生部(457天),其次是 Swissmedic(434天)、TGA(330天)和 EMA(37天)。相比阻碍,PMDA 呈现出独特的反向趋势,百强企业的申报间隔中位数显著增加,而非百强企业则大幅减少。这在一定程度上反映了2025年日本本土制药企业(通常占非百强企业的较大比例)批准比重的增加,拉低了该群体的整体申报滞后时间。
此外,CIRS特别关注了患者体验数据(PED)。虽然 2025 年 FDA NAS 批准中包含/考量PED的比例下降至 72%(2024 年为 84%),但其水平仍与前几年大体一致。考量多种 PED 来源(不仅是申办者提交的数据)的比例从 2021 年的 12% 增加到 2025 年的 20%,显示出审评中整合患者证据的手段更加主动和多样化。
识林®版权所有,未经许可不得转载
识林网址:www.shilinx.com





